百合AGPase基因RNAi載體構建及遺傳轉化研究
2018-08-04 01:34:02張進忠孫嘉曼李朝生劉挺燕范燕萍
西南農業學報 2018年6期
張進忠,孫嘉曼,李朝生,劉挺燕,范燕萍
(1. 華南農業大學林學與風景園林學院,廣東 廣州 510642;2. 廣西農業科學院生物技術研究所,廣西 南寧 530007;3. 廣西作物遺傳改良生物技術重點開放實驗室,廣西 南寧 530007)
【研究意義】食用百合地下鱗莖富含淀粉,是人們青睞的食材。百合鱗莖的形成、發育、生長與其淀粉、糖的合成與分解代謝密切相關[1],其繁殖過程依賴于鱗片再生種球,更依賴于淀粉的合成代謝,作為繁殖材料,鱗莖種球的膨大發育與淀粉的合成積累成正相關[2-3];在淀粉合成關鍵酶中,腺苷二磷酸葡萄糖焦磷酸化酶(AGPase)被認為是淀粉合成的限速酶[4],該酶與尿苷二磷酸葡萄糖焦磷酸化酶(UGP)酶偶聯,形成了蔗糖經萄糖-1-磷酸(G-1-P)形式轉化為腺苷二磷酸葡萄糖(ADPG)的合成路線,所生成的ADPG做為淀粉合酶的底物形成糖苷鍵延長合成淀粉。因此,AGPase對于以淀粉為收獲物的作物非常關鍵,對其基因的作用機理和調節模式進行相關研究,探索出能提高生物淀粉產量、改善淀粉品質的技術有利于農業生產。【前人研究進展】AGPase基因表達與生物淀粉的合成積累已有相關研究,一般認為其在植物體內過表達能顯著提高淀粉合成速率與含量,而反義調節則顯著下調其合成[5-7],然而對其具體作用的分子機理雖有報道,但仍知之甚少。在AGPase蛋白酶的多態性研究結果表明不同作物的AGPase酶存在多態性,所執行的功能各異,但都與淀粉合成有關;通過測序發現不同類型馬鈴薯中存在5種類型的AGPase酶,當酶為ISQV類型時,淀粉合成明顯高于其他拷貝類型[8];岳向文等[9]通過凝膠電泳鑒定出了60個小麥品種5種AGPase基因型,其中AGPabcd基因型酶活與淀粉含量最高,且各基因型具有不同的遺傳效應,AGPase大亞基的某些單核苷酸多態性(SNP)與穗粒數和千粒重密切相關[10];在對AGPase與木薯淀粉積累關系的研究中發現,木薯AGPase具有4種同工酶,其中AGPa、AGPb、AGPc基因型能顯著影響木薯塊根的淀粉積累[11];在水稻AGPase酶研究中發現了7種同工酶基因型,其中AGPS2b、AGPL2、AGPL3表達較強,是水稻胚乳中AGPase基因表達的主要形式[12]。涉及AGPase其他功能的研究目前較多。RNAi是研究基因功能、探索基因表達信號傳導通路的重要工具,在多種作物上進行基因功能的鑒定及創制新種質研究中發揮著重要作用。其通過RNAi沉默HMA2基因,闡明其介導鎘長距離運輸在馬藺適應重金屬鎘污染環境中的作用機制[13],構建沉默木薯eIF4E7基因的干擾載體,使木薯褐色條斑病毒與烏干達木薯褐色條斑病毒侵染時缺少翻譯起始因子,從而達到抗病毒的目的[14];通過RNAi過抑制大豆內源GmPEPc基因表達,增加油脂積累,獲得高油的轉基因大豆新品種[15],這些表明RNAi技術在研究基因功能和創新抗逆性、高品質種質有巨大的潛力。【本研究切入點】食用百合淀粉合成酶類基因與百合繁殖方式——鱗莖形成、膨大發育密切相關,而目前關于此類基因的研究非常少。【擬解決的關鍵問題】利用Gateway基因沉默技術,通過構建保守區域干擾表達載體并轉化植株,探索AGPase轉錄后水平的表達情況,為該基因功能的進一步研究及今后百合組培鱗莖膨大繁育基因調控技術的開發提供思路。
1 材料與方法
1.1 供試材料
1.1.1 植物材料 蘭州百合(Liliumdavidiivar.unicolor)組培苗,由廣西農業科學院生物技術研究所提供。
1.1.2 菌株與質粒載體 農桿菌GV3101(pMP90RK)感受態,大腸桿菌DH5α;入門載體pDONR221,目的載體pJawohl8-RNAi,具有AGPase基因(登錄號:KP751443)的pMD18-T克隆質粒由本實驗室保存。
1.1.3 主要試劑 GatewayTMBP ClonaseTMII Enzyme mix,規格100T,貨號11789100(Invitrogen);GatewayTMLR ClonaseTMEnzyme mix:規格20T,貨號11791019(Invitrogen);TaqDNA聚合酶、限制性內切酶購自TaKaRa,RNA提取試劑Trizol購于Invitrogen;逆轉錄試劑RevertAid First Strand cDNA Synthesis Kit:規格20T,貨號K1621(Thermo Scientific);DNA凝膠回收試劑盒AP-GX-50購于Axygen;PrimeScript RT Master Mix(Perfect Real Time)、SYBR Premix ExTaqTMII(Perfect Real Time)購于TAKARA;其余常規試劑為進口或國產分析純。
1.2 試驗方法
1.2.1 目的序列的擴增 根據基因序列,選擇中間300 bp保守區域作為干擾片段,利用Primer 5.0設計引物,引物由上海生工生物技術有限公司合成,其中通用引物為:
TF:5′-GGGGACAAGTTTGTACAAAAAAGCAGG CT-3′
TR:5′-GGGGACCACTTTGTACAAGAAAGCTGG GT-3′
含部分attB接頭的基因特異性引物為:
F:5′-AAAAAGCAGGCTTGAAGGAATTCggtaccG AGAACCCCAACTGGTTTCAGGG-3′,酶切位點Acc65I;
R:5′-AGAAAGCTGGGTTGCGGCCGCActcgag G GGGTATCAACCTTCATTGCCTTC-3′,酶切位點XhoI。
以帶有目的片段的質粒為模板,加含有部分attB接頭的基因特異性引物(F、R),進行第1輪PCR,然后以第1輪PCR產物為模板,加入通用引物,進行第2輪PCR。
PCR反應程序如下:將下列試劑于冰盒中溶解,并按以下次序將各組分在PCR管內混勻:模板1 μl,10×Buffer 5 μl,2 mM dNTP 5 μl,正反向引物各4 μl,25 mM MgCl23 μl,Taq1 μl,ddH2O 26 μl。擴增程序如下:94 ℃ 3 min;94 ℃ 30 s,60 ℃ 30 s,72 ℃ 1 min,30個循環;72 ℃ 10 min。1 %瓊脂糖凝膠電泳檢測PCR產物,回收擴增的PCR產物并純化。
1.2.2 構建入門克隆(將PCR產物構建進入門載體中) ①BP重組反應。反應體系組分如下:attB-PCR產物5 μl(>10 ng),pDONR221(150 ng/μl)1 μl,5×BP Clonase enzyme mix 2 μl,TE Buffer 4 μl。反應體系放于25 °C下溫育10 h,反應終止后,加2 μl的Proteinase K(2 μg/μl)在37 °C下處理10 min。反應產物應為含有目的片段的入門克隆pDONR-AGPase。②轉化。取100 μl DH5α感受態細胞,加入至上述反應體系中,冰上30 min,然后42 ℃水浴熱擊90 s,迅速放于冰上2 min,涂布到含有氨芐青霉素的LB固體培養皿上,37 ℃培養15 h。③PCR法篩選重組克隆。將LB平板倒置,常溫下隨機挑選平板上的陽性克隆并編號。每個PCR管里分裝16 μl ddH2O和正反向載體引物1 μl(5 pmol/μl),用槍頭在LB平板上輕點取轉化菌,然后將沾有菌體的槍頭置于相應的PCR管中吹打數次使其混合;將PCR其他組分配成PCR mix分裝于每個PCR管中,總體積達到25 μl,然后混勻,蓋緊管子。PCR mix配方如下:10×TaqReaction Buffer(Mg plus)2.5 μl,dNTP(10 mM)0.5 μl,Taq(5 U/μl)0.2 μl,ddH2O 4.8 μl。將混有菌體的PCR混合物短暫離心后置于PCR儀中,PCR反應程序如下:94 ℃,5 min;94 ℃ 30 s,58 ℃ 30 s,72 ℃ 30 s,25個循環;72 ℃ 10 min。凝膠電泳檢測PCR產物。
1.2.3 構建干擾表達載體(將入門克隆中的目的基因轉移至目的載體) ①LR重組反應。將下列試劑于冰盒中溶解,并按以下次序將各組分在1.5 mL離心管內混勻(總體積20 μl):pDONR-AGPase(≥100 ng/μl)3 μl,LR ClonaseTMReaction Buffer 4 μl,5×LR ClonaseTMenzyme mix 4 μl,pJawohl8-RNAi(≥300 ng/μl)1 μl,ddH2O 8 μl。移液槍混勻后并短暫離心,置于25 ℃下孵育過夜。加2 μl的Proteinase K(2 μg/μl)在37 °C下處理10 min。反應物應為含有正反向AGPase片段且中間有內含子的干擾表達載體:pJawohl8-RNAi-AGPase,取5 μl用于轉化。②轉化。取100 μl DH5α感受態細胞,加入至上述反應體系中,冰上孵育30 min,然后42 ℃水浴熱擊90 s,迅速回放于冰上2 min,涂布到含有氨芐青霉素的LB固體培養皿上,37 ℃培養15 h。③PCR法篩選重組克隆。將LB平板倒置,常溫下隨機挑選平板上的陽性克隆并編號。每個PCR管里分裝16 μl ddH2O和正反向載體引物1 μl(5 pmol/μl),用槍頭在LB平板上輕點取菌;將沾有菌體的槍頭置于相應的PCR管中吹打數下。將PCR其他組分配成PCR mix分裝于每個PCR管里,使總體積達到25 μl,然后混勻,蓋緊管子。PCR mix配方同上。將混有菌體的PCR混合物短暫離心后置于PCR儀中,PCR反應程序如下:94 ℃,5 min;94 ℃ 30 s,58 ℃ 30 s,72 ℃ 30 s,25個循環;72 ℃ 10 min。電泳檢測PCR產物,選擇出含目的片段的陽性克隆,再進行Acc65I、NheI與SpeI酶切鑒定。④重組質粒測序。挑選陽性克隆接種到5 mL LB/含氨芐青霉素的液體培養基中,37 °C搖床培養15 h,提取質粒送測序。
1.2.4 干擾載體轉化農桿菌 取出農桿菌GV3101感受態細胞于冰上凍融;加2 μl所構建的干擾載體質粒于100 μl感受態細胞中,用槍頭輕輕攪拌混勻;取出細胞與質粒的混合物轉入電擊杯中,在2500 V高壓下電擊;取出電擊杯,加入500 μl預冷的YEB培養液,輕輕吹打混勻,吸出菌液轉入1.5 mL離心管中,28 ℃,200 r/min振蕩培養5 h。取30 μl轉化的菌液涂在含相應抗生素的YEP培養基平板上(含50 mg/L卡那霉素、50 mg/L利福平),28 ℃避光倒置培養2 d后長出菌落,用接種環挑取單菌落接種于YEB液體培養基中(含50 mg/L卡那霉素、50 mg/L利福平),震蕩過夜培養。用特異引物進行菌液PCR檢測陽性克隆,并擴大培養后-80 ℃保存備用。
1.2.5 遺傳轉化和篩選 取培養好的百合愈傷組織,用鋒利手術刀片將愈傷組織切成0.5 cm2的小塊(帶有傷口),備用。取培養過夜的含有干擾載體的GV3101菌液,4000 r/min,室溫離心10 min,用MS鹽溶液重新懸浮菌體,使用時用MS鹽溶液稀釋至原體積的20倍。將準備好的組培塊在菌液中侵染10 min后取出,無菌濾紙吸去植物材料表面菌液,轉移到鋪有一層無菌濾紙的MS培養基上,28 ℃暗培養,共培養3 d。共培養后將材料轉移到分化培養基(MS+6-BA 0.3 mg/L+NAA 0.05 mg/L)上培養,28 ℃,光周期8L∶16D,直至長出新苗。
1.2.6 轉化植株的熒光定量PCR檢測 以轉化空質粒農桿菌GV3101的百合苗為對照,熒光定量PCR檢測經農桿菌GV3101轉化的含有AGPase干擾載體的植株AGPase基因相對表達量;取各處理的葉片下部膨大組織,使用TRIzol Reagent試劑盒抽提植物組織mRNA,使用PrimeScript RT Master Mix將mRNA擬轉錄成cDNA,特異性引物為:
18s F: 5′-TTCATATCACGTGCTGCATGG-3′
18s R: 5′-AGACGACTTCGGTGAGACG-3′
AGPase-F: 5′-GAGAACCCCAACTGGTTTCAG-3′
AGPase-R: 5′-GGTATCAACCTTCATTGCCTT-3′
以18s rRNA作為內參,使用SYBR Green熒光定量PCR試劑盒擴增目的片段和18s rRNA,反應條件設置為:95 ℃變性5 min;94 ℃ 30 s,57 ℃ 30 s,72 ℃ 30 s,40個循環。計算各基因的△CT,并采用2-△△CT法計算mRNA轉錄倍數,3次重復;采用SPSS 19.0 進行t檢驗統計分析。
2 結果與分析
2.1 目的干擾序列克隆
經過比對,選定的序列是AGPase(登錄號:KP751443)基因保守區域300 bp序列;圖1泳道1和2顯示的是經過兩輪PCR擴增出來的片段條帶,該條帶約為400 bp(含attB接頭),符合預期大小,表明擴增出了所選的AGPase基因干擾片段。
2.2 AGPase RNAi片段裝入到入門載體后菌落PCR結果
將所擴增的含有attB接頭的干擾片段與入門載體pDONR221進行BP反應,發生特異性位點重組形成入門克隆pDONR-AGPase與副產物。圖2為所設計引物對BP反應后的菌液PCR結果,其泳道1、2具有400 bp的清晰條帶,表明入門載體pDONR-AGPase已構建成功。
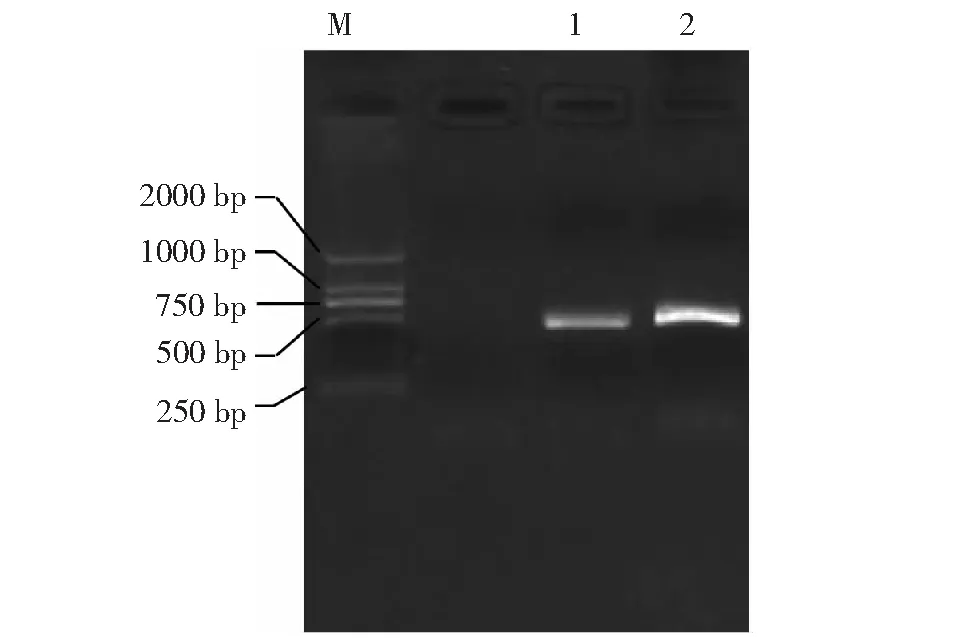
M:分子量標記;1~2:目的序列M: Marker; 1-2: Target sequence圖1 目的基因AGPase干擾片段Fig.1 The interference fragment of target gene AGPase
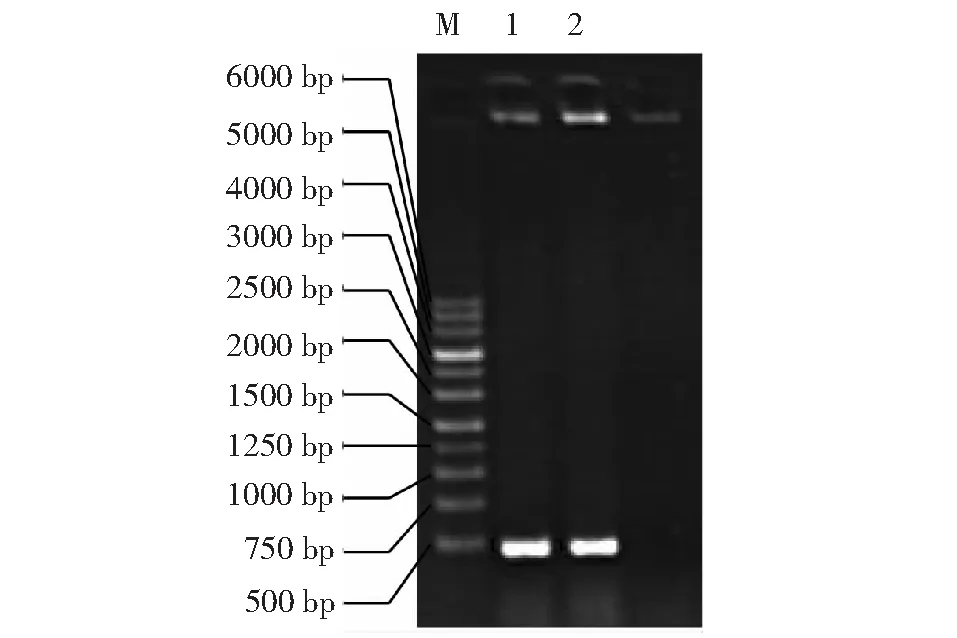
M:分子量標記;1~2:目的序列M: Marker; 1-2: Target sequence圖2 BP反應后菌液PCR結果Fig.2 PCR results of bacterial solution after BP reaction
2.3 入門載體pDONR-AGPase與目標載體pJawohl8-RNAi進行LR反應
入門載體pDONR-AGPase與目標載體pJawohl8-RNAi的LR反應后轉化大腸桿菌培養,對菌液進行PCR驗證,結果顯示擴增出了400 bp的條帶(圖3泳道1、2),與預期相符,表明經過LR反應,干擾序列成功轉入到目的載體,形成了所需的干擾表達載體pJawohl8-RNAi-AGPase。
2.4 干擾表達載體pJawohl8-RNAi-AGPase酶切結果
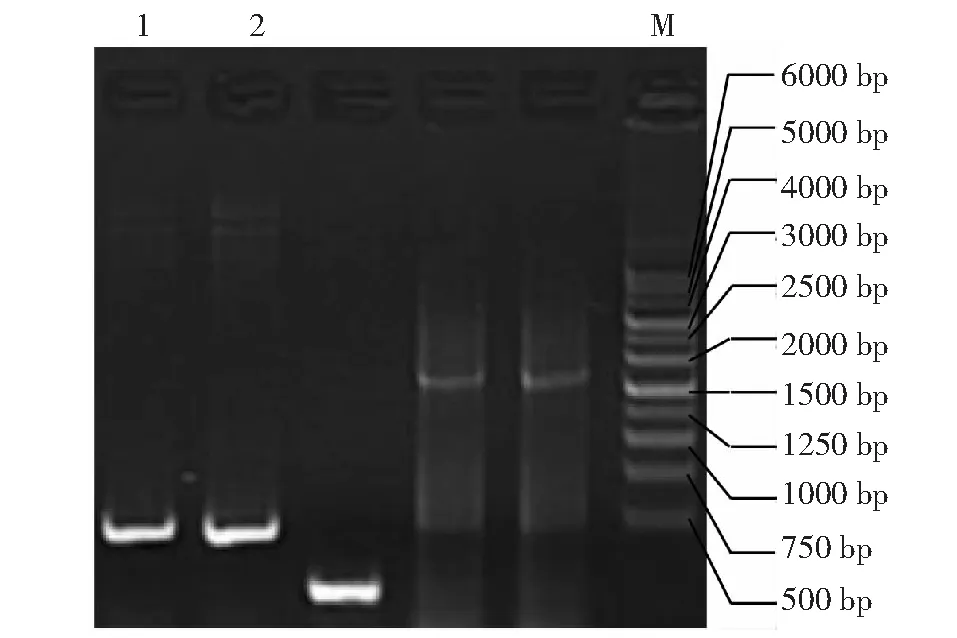
為進一步驗證表達干擾載體pJawohl8-RNAi-AGPase成功構建,選擇重組質粒載體上Acc65I、NheI與SpeI酶切驗證。如圖4所示,泳道1為pJawohl8-RNAi目的載體9019 bp;泳道2為pJawohl8-RNAi的NheI與SpeI雙酶切,顯示有約4300與4700 bp 2個條帶,NheI在質粒的8084 bp處,SpeI在質粒的3803 bp處,計算結果與條帶大小相符合,因其大小太相近,兩條帶接近在一起;泳道3為pJawohl8-RNAi的Acc65I酶切,因該載體沒有Acc65I酶切位點,所以還是呈現與泳道1大小一樣的條帶;泳道4為構建好的pJawohl8-RNAi-AGPase干擾表達載體,其大小約6540 bp;泳道5為pJawohl8-RNAi-AGPase干擾表達載體NheI與SpeI雙酶切結果,考慮到上述NheI與SpeI酶切位點位置,所以應具有約4280 bp與2260 bp條帶,泳道5條帶與此相符合;泳道6為pJawohl8-RNAi-AGPase干擾表達載體Acc65I酶切結果,應具有1026 bp與5514 bp條帶,結果與預測一致;泳道4、5、6的結果表明了pJawohl8-RNAi-AGPase干擾表達載體構建成功。圖5為最終成功構建的pJawohl8-RNAi-AGPase質粒圖譜。
M:分子量標記;1~2:目的序列M: Marker; 1-2: Target sequence圖3 LR反應后菌液PCR結果Fig.3 PCR results of bacteria solution after LR reaction

M:分子量標記;1:目的載體pJawohl8-RNAi;2:目的載體pJawohl8-RNAi的NheI與SpeI雙酶切;3:目的載體pJawohl8-RNAi的Acc65I酶切;4:干擾表達載體pJawohl8-RNAi-AGPase;5:干擾表達載體pJawohl8-RNAi-AGPase的NheI與SpeI雙酶切;6:表達干擾載體pJawohl8-RNAi-AGPase的Acc65I酶切M: Marker; 1: Destination vector pJawohl8-RNAi; 2: Digestion of pJawohl8-RNAi vector by NheI and SpeI enzyme; 3: Digestion of pJawohl8-RNAi vector by Acc65I enzyme; 4: Expression vector pJawohl8-RNAi-AGPase; 5: Digestion of expression vector of pJawohl8-RNAi-AGPase by NheI and SpeI enzyme; 6: Digestion of pJawohl8-RNAi-AGPase vector by Acc65I enzyme圖4 干擾表達載體pJawohl8-RNAi-AGPase酶切結果Fig.4 Digestion of interference vector pJawohl8-RNAi-AGPase

圖5 最終構建的pJawohl8-RNAi-AGPase質粒圖譜Fig.5 Constructed pJawohl8-RNAi-AGPase plasmid map
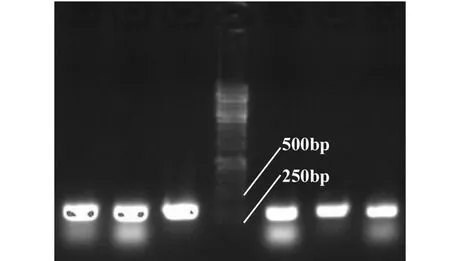
M:分子量標記;A:pJawohl8-RNAi-AGPase干擾質粒;AG:轉化到農桿菌的干擾質粒M: Marker; A: Interfering plasmid pJawohl8-RNAi-AGPase; AG: Interfering plasmid translated into Agrobacterium圖6 干擾片段的檢測Fig.6 Detection of interference fragments
2.5 干擾質粒轉化農桿菌PCR鑒定
通過電擊法將構建好的表達干擾載體轉入感受態農桿菌GV3101中,經過Kan、Rif、Amp抗性篩選,只有轉化菌株才能生長,進一步對轉化菌株進行PCR檢測;用AGPase干擾片段的引物(AGPase-F,AGPase-R)PCR擴增干擾質粒和轉化到農桿菌干擾質粒,以檢測所構建的pJawohl8-RNAi-AGPase干擾質粒是否轉化到農桿菌,并獲得陽性克隆。圖6所示A為pJawohl8-RNAi-AGPase陽性對照,AG為提取轉化的農桿菌質粒進行PCR,都具有300 bp大小的條帶,與所克隆的干擾片段大小相符合,表明構建的干擾質粒成功轉化到農桿菌GV3101中。
2.6 熒光定量PCR檢測AGPase基因轉錄后抑制
提取轉化植株RNA反轉錄為cDNA進行熒光定量PCR檢測,以比較轉化植株與非轉化植株AGPase基因相對表達量(圖7),結果顯示轉化含干擾質粒菌株的植株AGPase相對表達量大幅度降低,為對照的27.2 %。表明轉入的干擾片段對其轉錄的mRNA發生沉默作用。
3 討 論
本研究前期研究表明蔗糖能促進百合小鱗莖膨大發育,此處蔗糖作為百合生長利用的碳源具有以下作用:第一,作為合成淀粉的原料;第二,作為信號因子調控AGP相關啟動子表達,從而正調控AGP,AGP上調進一步提高了淀粉的合成。此外,植物激素在對AGP調控中也起到了非常重要的作用。AGPase基因的直接轉錄激活子尚未確定,Nagata等[16]從擬南芥cDNA中分離出轉錄因子AtWRKY20,通過粒子轟擊瞬時表達AtWRKY20,可增強ApL3啟動子的表達,啟動編碼一個蔗糖誘導的AGPase大亞基基因。AtWRKY20還激活編碼甘薯AGPase小亞基基因的ibAGP1啟動子。在AGPase蛋白結構研究中發現一個橫跨Pro103-Arg115的區域與AGPase酶底物結合及變構部分移動強烈相關,動態網絡路徑分析表明激活劑結合殘基(Lys39)及ATP的主要傳達通路與Pro103-Arg115殘基環有關,分子動力學表明Pro103-Arg115環在AGPase酶的變構反應中起著重要的作用,此為一個只存在于變構調節的糖核苷酸焦磷酸化酶中的獨特元件,可能是連接變構和催化位點觸發機制的一個關鍵部分[17]。通過單點突變研究發現大腸桿菌AGPase生理負性和正性變構調節劑分別為腺苷-5-單磷酸腺苷(AMP)和果糖-1,6-二磷酸(FBP),并由此提出了AGPase蛋白酶催化活性調控模型[18],并發現了一個與活性位點保持信息通路的傳遞變構信號的關鍵區域[19],這對理解AGPase變構調節具有重要意義。越來越多的研究證據闡述了AGPase的多態性、調控信息通路、分子結構變構調節與功能的關系等,本研究基于AGP響應于植物激素、碳源等因子調控作用下探索其功能研究的反向遺傳學方法,為下一步開展相關調控因子在百合鱗莖發育中的調控機制研究提供技術。

O:轉化含有空質粒農桿菌的植株;A:轉化含有AGPase干擾質粒農桿菌的植株O: Transformation of plant containing Agrobacterium; A: Transformation of plant containing Agrobacterium transformed with interfere plasmid圖7 轉化植株AGPase基因相對表達量Fig.7 Relative expression of AGPase Gene in transformed plants
Pinto等[20]利用RNAi將水稻黃斑病毒復制酶基因導入水稻培育出具有黃斑病毒抗性的水稻;柴曉杰等[21]采用 RNAi 技術能有效下調玉米支鏈淀粉含量。Gateway技術利用位點特異性重組原理實現片段定向轉移而得到方向正確的重組,其構建RNAi載體相對于傳統的酶切連接構建載體有方便、快捷、高效等優勢[22],只需獲得帶有目的干擾片段的入門克隆,通過LR反應就可將目的片段轉入所需要的表達載體中,滿足研究不同表達載體在不同生命體中的表達特征及目的基因的功能驗證。本研究采用兼容Gateway的植物基因沉默雙元載體pJawohl8-RNAi質粒帶有280 bp的內含子,將干擾序列分別正反向連接在兩邊,可有效表達dsRNA,形成siRNA實現基因沉默,所帶有的抗除草劑草銨膦抗性基因,有利于轉化后對轉基因植株的篩選,為研究該基因功能提供了材料與技術。
4 結 論
本研究選擇AGPase保守序列作為干擾片段,通過Gateway技術成功構建pJawohl8-RNAi-AGPase表達載體,并對百合愈傷組織進行遺傳轉化,誘導出AGPase轉錄后表達量下調的轉化植株。研究百合淀粉合成代謝途徑中AGPase基因的調節功能,為該基因功能的進一步探究及今后百合組培鱗莖膨大繁育基因調控技術的開發提供了材料與思路。
