FOXG1基因新發突變致先天性Rett綜合征變異型一例報告
2018-08-29 01:58:52牛巖趙澎蔡春泉舒劍波
天津醫藥 2018年8期
牛巖,趙澎△,蔡春泉,舒劍波
Rett綜合征主要表現為早期神經發育的退化,累及運動、認知及社交能力,常有刻板運動和癲癇發作。FOXG1(fork head box protein G1)基因突變所致的先天性Rett綜合征變異型是本病較為罕見的一型,臨床表現與其他突變相比有其特殊性,且目前尚無有效治療手段。我院收治了1例臨床確診病例,對其基因型與臨床表現進行分析,為臨床診斷此疾病提供參考。
1 病例報告
患兒 女,11個月齡,因自幼精神運動發育遲緩,于2016年5月16日就診于我院。患兒系胎1產1足月陰道分娩,出生體質量2.9 kg,其母親否認孕期異常及圍生期窒息史。患兒自幼精神運動發育遲緩,就診時豎頸仍欠穩定,不會翻身;運動、言語、認知、社交能力嚴重落后于同齡兒水平。患兒吃輔食較少,自入院前3個月至入院期間,母乳量不足,體質量增長緩慢,僅增加800 g;睡眠時間短,易醒;喜歡吃手,喜愛摸索衣領、被褥等物,經常伸舌;未報告可疑的癲癇發作事件。患兒父母體健,非近親結婚,家中無類似病史。入院時體格檢查:頭圍40.5 cm,前囟已閉,略見連眉,鼻頭稍圓,硬腭弓高,小下頜,目光不注視,呼之不應,不能逗笑。可見水平眼震,顱神經查體未見明顯異常,頸軟。四肢主動活動少,雙上肢肌張力減低,下肢肌緊張不全,雙側膝腱反射活躍,雙側巴氏征陰性。雙手中線位活動可,能互握,但主動抓物意識差,頻繁吃手,見刻板樣摸索動作。俯臥位僅能雙肘支撐挺身,輔助翻身欠靈活,扶坐不能,疊腰狀。扶站時雙下肢過伸、尖足。輔助檢查:血生化指標大致正常。血液氨基酸譜、酯酰肉堿譜及尿液有機酸分析未見明顯異常。染色體核型46,XX。神經電生理檢查:視覺誘發電位右側潛伏期大致正常,波幅降低,左側正常;體感誘發電位示四肢皮層段異常;聽覺誘發電位、四肢肌電圖、神經電圖未見異常。Gesell量表提示各測試功能區發育商嚴重落后于正常同齡兒水平:適應性27,粗大運動19,精細運動24,語言22,個人?社交10。國際評分系統(International Scoring System,ISS)評分[2]23分。腦電圖(EEG):主要異常為雙側枕區可見稍多散發中幅尖波(圖1)。頭顱磁共振成像(MRI)主要表現:額頂葉腦回發育不良,胼胝體細小(圖2)。基因測序:獲得患者家屬知情同意后,采集患兒及其父母外周血2 mL[枸櫞酸鉀(EDTA)抗凝],提取基因組DNA,采用靶向基因捕獲二代測序法對遺傳代謝病進行測序,并對患兒及其父母進行一代測序驗證;同時應用CytoScan HD芯片行染色體微陣列分析(Chromosome Microarray Analyze,CMA)。結果顯示:位于14q12的FOXG1基因有c.506dupG,p.G169Gfs*286雜合突變,該重復突變可導致移碼,進一步引起蛋白質序列的異常以及功能的改變。經驗證其父母FOXG1基因均為野生型(圖3),CMA檢測未檢測到有臨床意義的拷貝數變異。
本例隨訪至今,目前29個月齡,僅能進食流質食物,且進食時間長,總量低,偶有嗆咳,便秘,大便2 d 1次。體格發育顯著落后于同齡兒水平,身高90 cm(<P10)、體質量12.5 kg(<P3)。可自主翻身,尚不能獨坐,可短距離腹爬,雙踝關節攣縮,背屈受限,未見脊柱及髖關節畸形。16個月齡時有1次無誘因強直?陣攣樣發作病史,發作間期腦電如前。追視及注意力較前稍改善,雙手有主動抓物意識,但不能完成抓握過程。
2 討論
Rett綜合征主要表現為早期神經發育的退化,累及運動、認知及社交能力,伴有刻板運動,常有癲癇發作。經典的Rett綜合征是X連鎖顯性遺傳,與MECP2(methyl?CpG binding protein 2)基因突變有關。另有一些變異的表現型與Rett綜合征非常相似,多為常染色體顯性遺傳,如先天起病型、嬰兒期癲癇起病型、語言保留型等,后經研究證實,其中一些變異型與其他基因的突變有關,如CDKL5(cyclin?dependent kinase?like 5)基因及 FOXG1 基因[1]。FOXG1基因在端腦結構的發育及形成過程中發揮重要作用,該基因編碼一種轉錄抑制蛋白,其表達局限于胎兒及成人的腦和睪丸,在腦發育過程中,限定可以形成端腦的區域,如端腦神經上皮、視網膜鼻側半以及視神經表達。FOXG1突變及其基因周圍的異常均可引起發育性腦功能障礙。該基因的缺失、突變或失活與先天性Rett綜合征變異型的發生相關[2]。與其他基因型相比,該表型突出表現為先天或早期起病、獲得性小頭、語言功能嚴重受損、注意力缺陷、社交障礙、癲癇、喂養困難、身高體質量發育落后、嚴重的刻板運動、手功能及整體精神運動發育遲滯[3]。

Fig.1 The awake EEG of the patient圖1 患兒清醒腦電圖

Fig.2 Brain magnetic resonance imaging of the child圖2 患兒頭顱磁共振成像
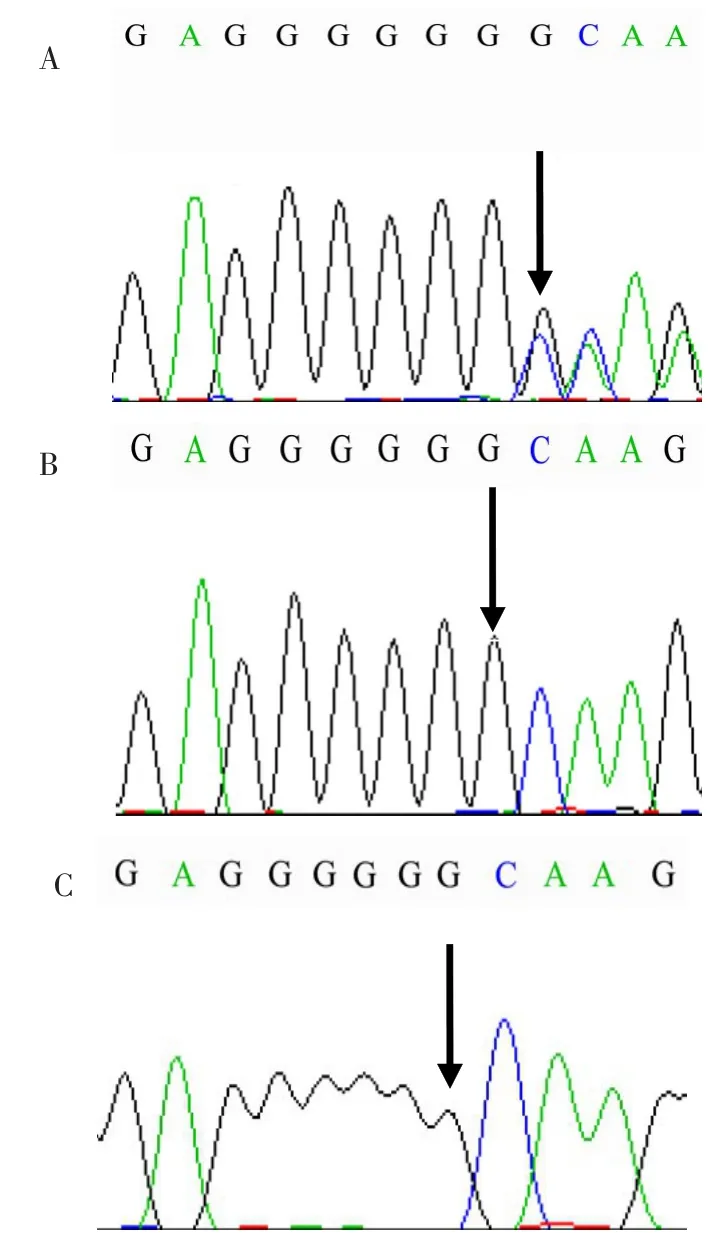
Fig.3 Genetic sequencing results of the patient and her parents圖3 患兒及其父母基因測序圖
以“Rett syndrome,FOXG1 mutation”或“Rett綜合征,FOXG1基因突變”為關鍵詞,對中國期刊全文數據庫(CNKI)、萬方數據知識服務平臺及美國國家生物技術中心(NCBI)、生物醫學文獻數據庫(Pubmed)自建庫至2018年2月發表論文進行檢索,共檢索到相關論文20篇,其中因包含混雜因素、病例資料不完善或不明確、部分論文及簡報資料不完整等原因,最終入選10篇。其中符合FOXG1基因突變且符合先天性Rett綜合征變異型臨床診斷病例,包含本例共19例(男2例,女17例),除2例[4,10]未獲得父母血樣及確切家族史證據外,其余病例均無顯性家族史,共發現19例FOXG1基因突變,其中9例為移碼突變,5例為無義突變,5例錯義突變,臨床表現詳見表1。
綜合上述文獻報道及本例患兒臨床表現,FOXG1基因突變所致19例Rett綜合征變異型,除1例[8]未詳細描述起病時間外,其余18例均在6個月齡以內起病;所有病例[4?13]均描述了確切的先天或者獲得性小頭,多數病例報道了額葉及胼胝體發育不良[4?12];同時,患兒可能存在睡眠障礙、注意力缺陷、刻板樣上肢運動,手功能障礙等表現。上述癥狀從臨床表型上體現了FOXG1基因在胎兒期及早期腦發育的重要意義[4]。除個別病例[9?12]外,大多數病例報道了不同程度、不同形式的癲癇發作,包括嬰兒痙攣樣發作[6,11]。在癲癇發作的預后方面,絕大多數FOXG1基因相關癲癇可通過3種以下抗癲癇藥物控制,以達長期無臨床發作狀態,預后較CDKL5基因突變病例輕[6]。除1例[6]外,其余患者均描述了明確的肌張力障礙癥狀,該患兒在7歲時可勉強行走,且穩定性差,其他患者均未獲得步行能力,少數長期隨訪病例顯示患兒持續處于臥床狀態[11?12]。吞咽過程中,口期及咽喉期相關肌肉多為隨意運動肌,亦存在相應的肌張力障礙。吞咽障礙是導致患兒體格發育障礙的主要原因,也是針對此類患者臨床工作的重要內容,應作為長期隨訪及診療方案的重要內容。多數病例[4,6?13]描述了吞咽、喂養困難、營養不良,部分大齡病例采用長期鼻飼、消化道造瘺等方式以改善整體營養狀態[9,12]。因長期隨訪病例較少,樣本中僅少數大齡病例報道了脊柱側突、足內翻以及關節攣縮的問題[9?12],提示在臨床工作中應注意早期預防相關骨骼畸形的發生。
對于FOXG1基因突變所致先天性Rett綜合征變異型,目前尚無有效的治療措施,在臨床確診后應進行綜合康復管理,以盡可能地提高患兒生存質量。具體內容包括:認知、言語、心理、運動等多方面整體康復;對癲癇發作、肌張力障礙等神經系統癥狀進行藥物干預;采取適當的方式改善喂養,從而改善營養狀態;注意早期預防骨骼畸形等。
本例患兒臨床表現主要為精神運動發育落后及倒退,頭圍小,癲癇發作和刻板的手部摸索動作,肌張力不全,額葉、胼胝體發育不良以及腦電圖異常等,ISS評分23分,符合先天性Rett綜合征變異型診斷標準[1]。基因測序結果提示FOXG1基因c.506dupG,p.G169Gfs*286雜合突變,該重復突變可致移碼,進而引起蛋白質序列異常及功能改變,其父母FOXG1基因均為野生型,考慮患兒為新發突變(de novo)。經查詢人類基因突變數據庫(HGMD)、Clinvar遺傳變異數據庫及dbSNP數據庫,該突變位點為新發突變,豐富了FOXG1基因突變譜。在臨床工作中,對于符合先天性Rett綜合征變異型表現的病例,建議行FOXG1基因突變檢查,對患者進行綜合康復管理,并對部分可預知的功能障礙進行預防性治療。
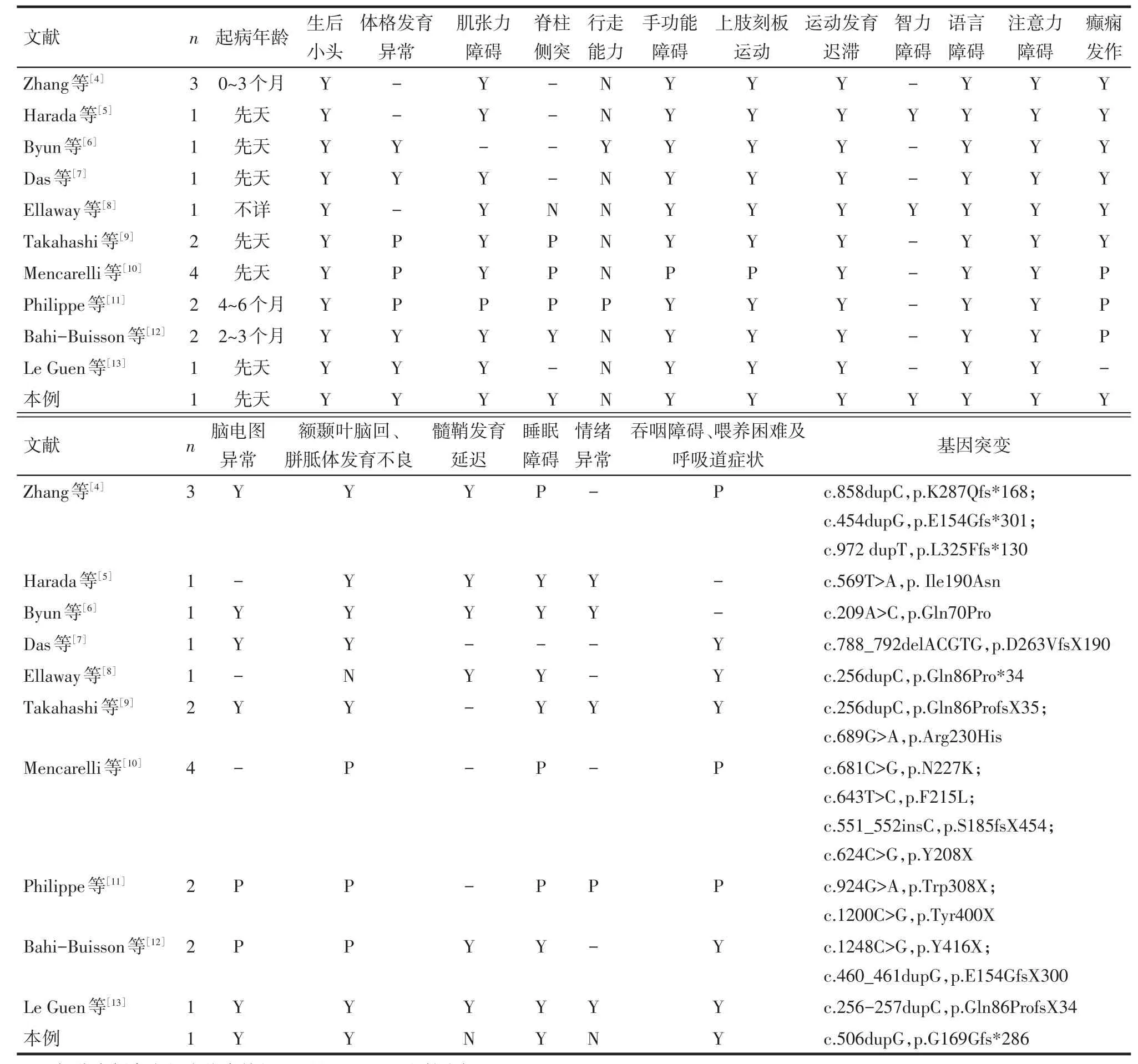
Tab.1 The clinical features of children with congenital Rett syndrome表1 FOXG1基因突變所致先天性Rett綜合征變異型主要臨床表現
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(2015年4期)2015-01-23 01:19:30
中國中醫藥現代遠程教育(2014年20期)2014-03-01 04:31:21
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
