以慢性腎功能衰竭為突出表現(xiàn)的新發(fā)致病突變非典型Bardet-Biedl綜合征1例報告
2018-08-30 08:57:10申金鳳帥蘭軍吳小川
中國臨床醫(yī)學(xué)
2018年4期
申金鳳,曹 艷,帥蘭軍,吳小川
中南大學(xué)湘雅二醫(yī)院兒童醫(yī)學(xué)中心,長沙 410011
1 病例資料
1.1 首次入院情況 患兒男性,11歲,因“多飲、多尿2年余,乏力半月余”于2015年3月31日第1次入住我科。患兒2013年上半年無明顯誘因出現(xiàn)多飲、多尿,每天飲水約2 200 mL,尿量達(dá)3 600 mL/d,伴乏力。于當(dāng)?shù)蒯t(yī)院查腎功能:尿素氮19.12 mmol/L、肌酐440.5 μmol/L;電解質(zhì)檢查:鉀1.86 mmol/L;血常規(guī):血紅蛋白76 g/L。腎臟彩超示:右腎囊腫。當(dāng)?shù)蒯t(yī)院初步診斷為“慢性腎炎”。既往史、個人史、家族史無特殊。入院查體:身高130 cm(低于134.5 cm兩個標(biāo)準(zhǔn)差),體質(zhì)量32 kg,體質(zhì)指數(shù)(BMI)18.9 kg/m2。發(fā)育落后,貧血面容,雙眼斜視;手指短,無杵狀指及多指/趾(圖1);睪丸容積約2 mL,陰莖短小。病理征陰性。入院后查血常規(guī):血紅蛋白96 g/L;尿常規(guī):尿蛋白(+)、尿糖()、尿隱血();腎功能:尿素氮21.06 mmol/L,肌酐389.3 μmol/L,肌酐清除率16.36 μmol/L(CKD 4期)。腎臟彩超:雙腎實質(zhì)彌漫性病變;腎臟MRI:雙腎多發(fā)小囊腫。
腎組織活檢:腎組織免疫熒光染色結(jié)果IgA(-)、IgG(+)、IgM(+/)、C1q()、C3(+)、C4(-)、Fb(+)。病理檢查結(jié)果(圖2):(1)C1q腎病(增生硬化型);(2)間質(zhì)纖維化。電鏡檢查結(jié)果:腎小球中度系膜增生,腎小管壞死或萎縮,間質(zhì)纖維增生。予潑尼松1 mg/kg口服治療,輔以補鈣、控制血壓等對癥治療,并于2015年4月19日帶藥出院。出院時患兒尿素氮25.58 mmol/L,血清肌酐445.5 μmol/L,尿量約2 100 mL/d。出院后規(guī)律服藥2個月后,自行停藥,予中藥(具體不詳)治療,病情逐漸加重。
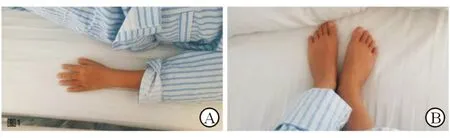
圖1 患兒指(A)、趾(B)表現(xiàn)
1.2 再次入院情況 2016年1月28日復(fù)查腎功能:尿素氮48.02 mmol/L,肌酐868 μmol/L。患兒伴發(fā)熱、咳嗽,腹脹,尿少,視物模糊、斜視,偶有胸悶、心悸等不適,遂再次入住我科。入院查體:身高131 cm(低于134.5 cm兩個標(biāo)準(zhǔn)差),體質(zhì)量37 kg,BMI 21.56 kg/m2。身材矮小,雙眼斜視,雙眼瞼輕度水腫,口腔潰瘍,伸舌居中、輕微顫動,咽紅,扁桃體Ⅱ°腫大,雙下肺呼吸音減弱,心律齊、無雜音,腹部膨隆、移動性濁音陽性。……
登錄APP查看全文
