Pd-Ag/Al2O3催化劑的制備及性能
2018-08-30 03:01:06衛(wèi)國賓趙曉瑋
石油化工 2018年8期
衛(wèi)國賓,穆 瑋,趙曉瑋
(1.中國石化 北京化工研究院,北京 100013;2.北京順安奇特氣體有限公司,北京 101300)
W/O微乳液是水在油中形成的微乳化體系,具有熱力學穩(wěn)定、澄清透明且各向同性的特點[1]。W/O微乳液由于具有獨特的微水池結(jié)構,被應用于金屬納米粒子的合成反應。Solanki等[2]在AOT(琥珀酸雙(2-乙基己基)酯磺酸鈉)、環(huán)己烷和水組成的微乳液體系中合成不同粒徑大小的納米Ag。張敬輝等[3]在納米SiO2的微乳液實驗中發(fā)現(xiàn)SiO2顆粒平均粒徑隨助表面活性劑分子鏈的增加逐漸變小。朱文慶等[4]通過改變微乳液體系的反應溫度,得到不同形貌的微/納米Sm2O3。
金屬元素的原子外層d電子層具有空軌道時,會表現(xiàn)出不同程度的對不飽和烴的化學吸附能力。20世紀50年代人們研究發(fā)現(xiàn)Pd在乙炔加氫反應中是最具活性的金屬,80年代開始雙金屬催化劑體系地開發(fā),并實現(xiàn)了Pd-Ag/Al2O3催化劑的工業(yè)化應用[5],活性組分Pd和助劑Ag在載體上呈蛋殼型分布,減少了傳質(zhì)阻力的影響[6]。Ag可以減少Pd粒子的比表面積,形成Pd-islands和具有單一晶面的Pd表面原子[7]。Pd外表面上點綴Ag,使相鄰的Pd原子數(shù)量減少,消弱了催化劑的化學吸附氫(解離)能力[8]。相應的也大大降低了那些不受歡迎的副產(chǎn)物地生成,如綠油和乙烷[9]。
本工作通過研究聚乙烯基吡咯烷酮(PVP)對聚氧乙烯(4)月桂醚(Brij30)/正辛烷/水微乳液體系的影響,合成了能更穩(wěn)定、均勻制備納米合金粒子的微乳液體系,將合成的Pd-Ag納米粒子成功負載在載體上,制備了Pd-Ag/Al2O3催化劑,采用SEM、H2-TPR及等溫H2變壓吸附等表征手段對不同方法制備的加氫催化劑進行表征,并考察了催化劑在乙炔加氫反應中的活性和選擇性。
1 實驗部分
1.1 試劑
Brij30:x(H2O)< 1×10-6,Sigma-Aldrich公司;正辛烷:EP,99%(w),Acros organicsh公司;PVP3(M= 3 500)、PVP8(M= 8 000):Acros organicsh公司;PVP10(M= 10 000):Sigma-Aldrich公司;99.9%(w)AgNO3、99.9%(w)Pd(NO3)2:Strem 公司;氧化鋁:AR,中國石化催化劑分公司;水合肼:50%(w),北京化工廠。
1.2 W/O型微乳液體系擬三元相圖的繪制
在25 ℃恒溫條件下,由Brij30/正辛烷組成乳化液,再逐滴加入水或PVP水溶液,體系將自發(fā)形成透明或半透明狀態(tài)。按Brij30與正辛烷的質(zhì)量比(0∶10,1∶9,2∶8,3∶7,4∶6,5∶5,6∶4,7∶3,8∶2,9∶1,10∶0)配成系列的乳液體系,測定最大增溶水量,確定W/O微乳液的穩(wěn)定區(qū)域,從而繪制微乳液擬三元相圖。
1.3 Pd-Ag/ Al2O3催化劑的制備
1.3.1 Brij30/正辛烷/水微乳體系制備催化劑
在25 ℃恒溫水浴下,將10 mL Brij30和14 mL正辛烷均勻混合。配制8 mL Pd(NO3)2和AgNO3混合水溶液加入至Brij30和正辛烷乳液中,攪拌20 min,形成穩(wěn)定的微乳液體系。
在攪拌過程中,向微乳液中逐滴加入還原劑水合肼溶液0.5 mL,攪拌4 h,保證還原反應的完成,溶液的顏色由淡黃色變?yōu)楹谏f明Pd-Ag納米粒子懸浮在溶液中。將含有Pd-Ag納米粒子的微乳液浸漬到100 g的Al2O3載體上,經(jīng)過110 ℃干燥2 h,550 ℃焙燒4 h后,在120 ℃下氫氣還原1.5 h后備用,Pd-Ag納米負載催化劑的Pd含量為0.02%(w),Pd/Ag摩爾比為1.5,標記為BO-H-13。
1.3.2 Brij30/正辛烷/PVP微乳體系制備催化劑
在25 ℃恒溫水浴下,將10 mL Brij30和14 mL正辛烷均勻混合。配制8 mL Pd(NO3)2和AgNO3混合的PVP水溶液并加入Brij30和正辛烷乳液中攪拌20 min,形成穩(wěn)定的微乳液體系。
在攪拌過程中,向微乳液中逐滴加入還原劑水合肼溶液0.5 mL,攪拌4 h,保證還原完成后將微乳液浸漬到100 g的Al2O3載體上,經(jīng)過110 ℃干燥2 h,550 ℃焙燒4 h后,在120 ℃下氫氣還原1.5 h后備用,Pd-Ag納米負載催化劑的Pd含量為0.02%(w),Ag/Pd摩爾比分別為0.5,1.0,1.5,2.0,分別標記為BO-P-21,BO-P-22,BOP-23,BO-P-24。
1.4 浸漬法制備催化劑
取 100 g Al2O3載 體 浸漬于 50 mL Pd(NO3)2和AgNO3混合水溶液中,浸漬5 h,然后過濾、干燥、焙燒、還原,制成Pd-Ag/Al2O3催化劑。制備Ag/Pd摩爾比為0.5,1.0,1.5,2.0的催化劑,分別標記為BC-H-11,BC-H-12,BC-H-13,BCH-14。
1.5 表征方法
采用美國FEI公司的XL-30型掃描電子顯微鏡對催化劑的表面和剖面的形貌進行表征,并進行元素能譜掃描;采用美國麥克儀器公司的AutoChem 2920型化學吸附儀對催化劑進行程序升溫還原和等溫H2變壓吸附分析,程序升溫范圍:-50~1 100 ℃,載氣為He,H2升溫速率為10 ℃/min。
1.6 催化劑的性能評價
碳二餾分選擇加氫反應在Amtech公司Spider型固定床反應器上進行,反應的氣態(tài)空速為12 000 h-1,原料氣組成(x)為:乙烷6.570%、乙烯92.239%、乙炔0.480%。催化劑床層的上、中、下部分別裝有熱電偶,檢測床層入口、反應段和出口處的溫度。原料氣和氫氣由上端并流進入反應器,氫炔摩爾比為1.5,氣體質(zhì)量流量計計量。采用Agilent公司6890型氣相色譜儀對加氫前后的碳二餾分進行定量分析,F(xiàn)ID檢測,柱溫50~60 ℃,采用面積歸一化法定量,炔烴檢出量最小為5×10-7。乙炔轉(zhuǎn)化率(X)和乙烯選擇性(S)分別由式(1)和式(2)計算。

2 結(jié)果與討論
2.1 PVP對Brij30/正辛烷/水微乳液體系的影響
制備金屬納米顆粒的微乳液體系中,金屬鹽只溶解于水相中,足夠大的增溶水量才能保證金屬鹽全部溶解于微乳液中。圖1為Brij30/正辛烷/水在不添加助表面活性劑條件下形成的W/O微乳液體系的三元相圖。由圖1可看出,Brij30/正辛烷/水體系在不添加助表面活性劑條件下可形成W/O微乳液。當Brij30為30.63%(w)、正辛烷為30.63%(w)時,該微乳液體系可以獲得的最大增溶水量為38.74%。
Brij30的水溶性來自于氧乙烯基(EO)鏈中醚氧原子與水分子形成的氫鍵作用。通過增加或減少親水的EO鏈長,可在很大范圍內(nèi)改變非離子表面活性劑的親水性,如果以EON 表示非離子表面活性劑分子中EO的平均數(shù),則隨EON的增加,親水作用增強,所形成的微乳體系的微結(jié)構將發(fā)生從WinsorⅡ→Winsor Ⅲ→WinsorⅠ的轉(zhuǎn)變,也就是說EON較低的非離子表面活性劑更有利于形成W/O微乳液體系。Brij30的EON為4,是芐澤類聚氧乙烯脂肪醇醚非離子表面活性劑中最低的。

圖1 Brij30/正辛烷/水體系的三元相圖Fig.1 Ternary phase diagram of the Brij30/n-octane/H2O system.
圖2是不同相對分子質(zhì)量的PVP的微乳液體系擬三元相圖。PVP相對分子量對W/O微乳液穩(wěn)定區(qū)域的影響較大。考慮到在微乳液中水滴粒徑都在納米范圍內(nèi),PVP在水滴中形成長鏈會纏繞在一起,因此本實驗中選用了相對分子質(zhì)量較低的PVP進行考察。從圖2可看出,隨表面活性劑Brij30含量的增大,受PVP10分子體積的影響,微乳穩(wěn)定區(qū)明顯縮小,甚至水溶液無法溶解在Brij30中;而Brij30/正辛烷/PVP3所形成的微乳液穩(wěn)定區(qū)域比Brij30/正辛烷/水體系穩(wěn)定區(qū)域略大。PVP3微乳液的最大增溶水量達40.03%,PVP10微乳液的最大增溶水量為34.96%。
微乳液中水相里鹽溶液的含量增加,離子強度增大,則界面層表面活性劑的親水基團彼此之間的斥力相對降低,導致液滴體積變小,部分水分子被擠出液滴,所以在固定溫度下W/O微乳液的最大溶量下降[10]。考慮到Pd和Ag的硝酸溶液在水中的溶解度和離子強度的影響,微乳液的最大增溶水量需達30%以上。PVP(M=3 500~10 000)的微乳液均可滿足制備Pd-Ag納米粒子的要求。

圖2 不同相對分子質(zhì)量的PVP的微乳液體系的三元相圖Fig.2 Ternary phase diagram of the Brij30/n-octane/PVP system with different molecular weights of PVP.
2.2 SEM表征結(jié)果
圖3是對干燥后沒有高溫焙燒的BO-P-22催化劑進行SEM和EDX的表征結(jié)果。在催化劑表面以灰色線段為軸,分別測定不同位置的Pd,Ag,O,C,Al元素的能譜。從圖3a可看出,催化劑表面Al和O元素沒有出現(xiàn)基線,說明其含量較高,Pd,Ag,C元素的能譜出峰位置接近,尤其是Pd和Ag的能譜峰出現(xiàn)位置完全相同,這被認為是Pd和Ag呈合金形式存在,且附著在有機物上。從圖3b可看出,沿著催化劑剖面由表層深入到內(nèi)部的灰色軸線上,Pd,Ag,C元素大量富集在催化劑表層5 μm以內(nèi),在剖面深處沒有明顯檢出各活性組分Pd和Ag的能譜。微乳液制備的催化劑呈蛋殼型分布在載體表層。后續(xù)焙燒和還原不會導致在表面富集的Pd-Ag合金向載體體相內(nèi)遷移,因為催化劑制備中影響活性組分殼層分布的主要因素來自于浸漬和干燥步驟。

圖3 BO-P-22催化劑表面(a)和剖面(b)的SEM照片及EDX的元素分布Fig.3 SEM images and element distributions of the surface(a) and the section(b) of BO-P-22 catalyst.
2.3 H2-TPR表征結(jié)果
圖4為浸漬法和微乳液法制備的催化劑的H2-TPR曲線。從圖4a可看出,BC-H-11,BC-H-12,BC-H-13,BC-H-14催化劑的低溫氫還原峰溫度分別為54,66,74,82 ℃,高溫氫還原峰溫度分別為377,403,420,440 ℃,而BC-H-12,BCH-13,BC-H-14在230,240,250 ℃還存在著氫還原峰。這說明浸漬法制備的催化劑中Pd的氧化形態(tài)較為復雜,隨Ag含量的增加,低溫和高溫的氫還原峰都向高溫方向移動。雖然Ag對Pd起稀釋作用,但部分Ag覆蓋在Pd的外露活性位,減弱了加氫能力,隨Ag含量增加,這種減弱效果更加顯著,降低了催化劑的加氫效率,因此還原難度增大。BC-H-11和BC-H-12在150 ℃附近都出現(xiàn)了明顯的負峰,這是由于Ag含量較低的情況下,體相Pd上存在著H2溢流現(xiàn)象,負峰是氫鈀酸(PdHx)的分解峰[11]。從圖4b可看出,BO-P-21,BO-P-22,BO-P-23,BO-P-24催化劑的低溫氫還原峰溫度為48,8,0,4 ℃,只有BO-P-21在390 ℃有高溫氫還原峰。以合金形式被負載到載體上后經(jīng)高溫焙燒所得到氧化態(tài)的Pd-Ag合金化合物物種均勻單一,且與載體之間的作用力較弱,氫還原峰絕大部分在0~10 ℃的低溫區(qū)。這說明微乳液比浸漬法制備的催化劑更容易還原,微乳液制備的Pd-Ag合金形態(tài)中,Ag對Pd的稀釋作用和電子效應更為明顯,對Pd的有效活性位消弱減小。BO-P-21催化劑中Ag含量低,微乳液形成的納米粒子中有純Pd粒子存在,在高溫焙燒后又形成氧化鈀形態(tài),因此低溫還原峰偏高,且在390 ℃出現(xiàn)明顯的高溫氫還原峰,在220 ℃左右也存在PdHx的分解峰。
2.4 催化劑的H2吸附等溫線
在乙炔加氫反應中,氫以兩種相態(tài)存在,一種是表面活性組分Pd解離吸附氫,另一種是體相中Pd吸收氫。氫分子吸附在Pd表面解離為兩個氫原子。一個表面Pd原子吸附一個氫原子。圖5為三種類型催化劑的氫吸附等溫線。從圖5可看出,單Ag催化劑有非常小的氫吸附量,實際上Ag原子不具備氫吸附能力,少量的氫吸附可能是氧化鋁的吸附造成的。單體Pd催化劑的吸附等溫線存在兩個平臺,分別對應中氫吸附和吸收兩種方式,第一個平臺(p=(13~33)×10-4MPa)主要是表面Pd的吸附氫為0.000 95 mmol/g。第二個平臺(p=(93~200)×10-4MPa)是體相中Pd的吸收氫為0.000 4 mmol/g。
BO-P-22催化劑僅僅只有一個平臺,吸附氫量為0.001 mmol/g,這應該全部來自活性組分表面解離吸附氫,PVP獨特的伸展性和吸附性,使Pd-Ag合金納米粒子附著在PVP上呈薄膜形態(tài)屬負載并分散在Al2O3載體表面,減少了載體表面活性組分的團聚現(xiàn)象,體相Pd大大降低。體相Pd更容易使乙炔深度加氫生成乙烷。

圖4 浸漬法(a)與微乳液法(b)制備的催化劑的H2-TPR曲線對比Fig.4 Comparison of H2-TPR spectra of the catalysts prepared by impregnation (a) and microemulsion (b).
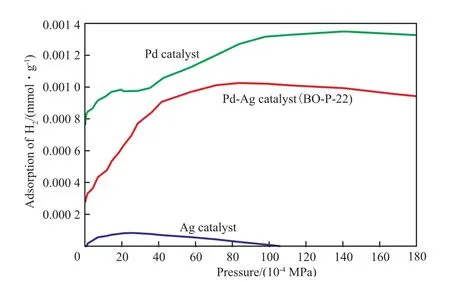
圖5 催化劑的氫吸附等溫線Fig.5 Isotherms of hydrogen adsorption on the catalysts.
2.5 不同方法制備的Pd-Ag/Al2O3催化劑的乙炔加氫性能
圖6為不同方法制備的催化劑乙炔加氫活性和選擇性隨溫度的變化曲線。從圖6可看出,三種催化劑的活性和選擇性隨著溫度變化規(guī)律相同,即溫度越高,活性越高,選擇性越低。在反應溫度達到140 ℃時,BC-H-14的選擇性為38%,在三個催化劑中下降幅度最大。BO-H-13和BO-P-22的選擇性在反應溫度達到100 ℃后,下降速度減慢,并出現(xiàn)平臺,選擇性可保持在50%以上,由此可以看出,微乳液制備的催化劑活性和選擇性都明顯高于傳統(tǒng)浸漬法制備的催化劑。
傳統(tǒng)浸漬法制備的碳二加氫催化劑中,當Ag/Pd摩爾比為2時,催化劑的催化性能最佳[12]。Brij30/正辛烷/H2O微乳液體系制備的催化劑中,最佳Ag/Pd摩爾比可降低至1.5[13]。Brij30/正辛烷/PVP體系制備Ag/Pd摩爾比為1.0的BO-P-22催化劑在各個溫度條件下的出口乙炔控制能力和乙烯選擇性都優(yōu)于其體系下制備的最優(yōu)催化劑,結(jié)果如表1所示。由于選擇加氫脫除乙炔為精制反應,在工業(yè)上往往要求乙炔被脫除到1×10-6(x)以下,因此評價中出口乙炔出現(xiàn)幾個或十幾個10-6(x)及以上的差別是可清晰地分清催化劑的性能優(yōu)劣的。

圖6 不同制備方法的催化劑的轉(zhuǎn)化率和選擇性Fig.6 Conversions and selectivities of catalysts preparedby the different methods of preparation.
采用不同制備方法得到的乙炔加氫催化劑都會有最佳的Ag/Pd摩爾比。當Ag/Pd摩爾比較低時,Ag對活性組分Pd的修飾作用不足,催化劑加氫選擇性差,大量的乙烯加氫會導致催化劑的乙炔最高轉(zhuǎn)化率偏低;當Ag/Pd摩爾比過高,催化劑的加氫活性偏弱,需提高反應溫度滿足乙炔加氫轉(zhuǎn)化率的要求,而反應溫度的升高會導致乙烯加氫和炔烴聚合速率增大,大量乙烷和綠油的生成,降低了乙烯收率。浸漬法制備的Pd-Ag/Al2O3催化劑表面多以單獨Pd和Ag粒子堆積為主,能對Pd起到協(xié)助效應的有效Ag偏低。Brij30/正辛烷/H2O體系制備互溶的Pd-Ag合金負載催化劑,不但節(jié)省Ag/Pd摩爾比,且控制出口乙炔的能力和乙烯選擇性進一步提升,由此表明合金形式的Ag對Pd協(xié)助作用更佳,Ag的有效利用率更高。Brij30/正辛烷/PVP體系制備的催化劑在不降低乙烯選擇性的條件下,能獲得更高的出口乙炔控制能力。說明微乳液中PVP的加入,能進一步提升Pd-Ag合金粒子的金屬互溶性,且粒度分布均一穩(wěn)定,此外利用PVP吸附和伸展性,使Pd-Ag合金納米粒子呈薄膜形態(tài)屬負載并分散在Al2O3載體表面,不會使金屬粒子從載體上脫落和聚集,從而進一步降低Ag/Pd摩爾比。

表1 不同制備方法的催化劑的出口乙炔和乙烯選擇性比較Table 1 Comparison on acetylene slip and ethylene selectivity of catalysts prepared by different methods
3 結(jié)論
1)在Brij30/正辛烷/水體系中加入相對分子質(zhì)量較低的PVP更有利于形成較大微乳液穩(wěn)定區(qū)域,Brij30/正辛烷/PVP(M= 3 500~10 000)體系都滿足制備Pd-Ag納米粒子的要求,可用于Pd-Ag合金納米粒子合成反應。
2)不同方法制備的催化劑相比,Brij30/正辛烷/PVP體系制備的Pd-Ag/Al2O3催化劑在乙炔加氫反應中活性和選擇性最佳,Ag/Pd摩爾比最低,大大提升了催化劑活性組分的加氫效率。
3)微乳液浸漬法中PVP的加入,可有效地規(guī)整了Pd-Ag合金粒子組成和形態(tài),并隨PVP薄膜形態(tài)屬負載并分散在Al2O3載體表面,有效抑制了單質(zhì)Pd的出現(xiàn)和聚集。
猜你喜歡
新世紀智能(數(shù)學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
應用化工(2014年3期)2014-08-16 13:23:50
