兒童擴張型心肌病的臨床特點及1例家族性病兒的基因突變分析
2018-09-26 10:48:14
精準醫學雜志 2018年4期
(廣西醫科大學第一附屬醫院兒科,廣西 南寧 530021)
擴張型心肌病(DCM)是兒童和青少年心源性猝死的常見病因[1]。兒童心肌病約占兒童心臟病的1%[2],DCM是兒童心肌病中最常見的類型,病因復雜,包括心肌炎、神經肌肉疾病、家族性心肌病、遺傳代謝病等[3],預后差,僅有30%~40%病兒可明確病因[4-5]。本研究分析了49例DCM病兒的臨床特點,并分析了1例家族性DCM的基因突變結果。現將結果報告如下。
1 資料與方法
1.1 一般資料
收集2006年1月—2016年1月于我院兒科首次住院治療的DCM病兒49例,男22例,女27例;起病年齡6個月~14歲,平均起病年齡為(89.2±49.2)個月;其中<1歲者4例,1~5歲者13例,>5歲者32例。
1.2 研究方法
所有DCM病兒均詳細詢問病史,體格檢查,收集病兒的年齡、性別、臨床癥狀、體征、肌酸激酶同工酶(CK-MB)、心肌鈣蛋白I(CTnI)、心電圖、胸片、超聲心動圖以及基因檢測結果,部分病兒行心臟CT或者心臟核磁共振檢查。入選標準:診斷符合1995年WHO/ISFC心肌病分類標準和中國心肌病診斷與治療建議工作組制定的心肌病診斷建議中的標準[6-7];年長兒心功能評估采用NYHA心功能分級法,嬰幼兒采用改良的Ross評分法。采集家族性DCM先證者、先證者父母、先證者妹妹、先證者哥哥的靜脈血,采用一代測序法(Sanger法)完成基因測序及變異位點的篩查,確證變異位點的參考數據庫為HGMD Pro、PubMed、1000Genomes和dbSNP。基因檢測和數據判讀由北京康旭醫學檢驗所完成。49例病兒出院后門診隨訪或電話隨訪,隨訪時間至2016年12月,以死亡或心臟移植作為隨訪終點。
1.3 統計學方法

2 結 果
2.1 DCM病兒的臨床基線資料
49例DCM病兒入院時主要的癥狀和體征為咳嗽、氣促、乏力、肝臟增大以及心音低鈍等,其中40例(81.6%)病兒心胸比增大,大部分病兒合并呼吸道感染,但無栓塞事件發生。見表1。
2.2 DCM病兒的心臟超聲檢查結果
49例DCM病兒左心室均增大,左心室舒張末期內徑(LVDd)平均值為(57.4±8.9)mm,左心室收縮末期內徑(LVDs)平均值為(47.6±8.3)mm,左心室射血分數(LVEF)<30%者16例(32.7%),左心室短軸縮短率(LVFS)<20%者31例(63.3%)。所有病兒均存在不同程度的瓣膜反流,但僅1例為重度瓣膜反流;25例(51.0%)病兒存在肺動脈高壓,其中僅1例為重度肺動脈高壓;20例(40.8%)病兒存在少量心包積液,3例(6.1%)病兒出現左心室附壁血栓。
2.3 DCM病兒的隨訪情況
病兒均給予強心劑、利尿劑和ACEI等治療,合并感染者給予抗感染治療。首次住院病兒無死亡者,出院后獲得隨訪32例,隨訪時間1~131個月,平均29.25個月,死亡13例,5例病兒出院后堅持規律服用強心和利尿藥半年~1年后停藥,但至隨訪時生活質量可,無明顯運動受限;14例病兒目前堅持規律用藥,用藥時間2~5年,日常活動無明顯影響;余17例失訪。13例死亡病兒均因心力衰竭進行性加重而死亡,無猝死者,無心臟移植者。Kaplan-Meier生存曲線顯示,病兒1、3和5年生存率分別為68.2%、54.4%和54.4%,診斷后2年內死亡風險最高。見圖1。
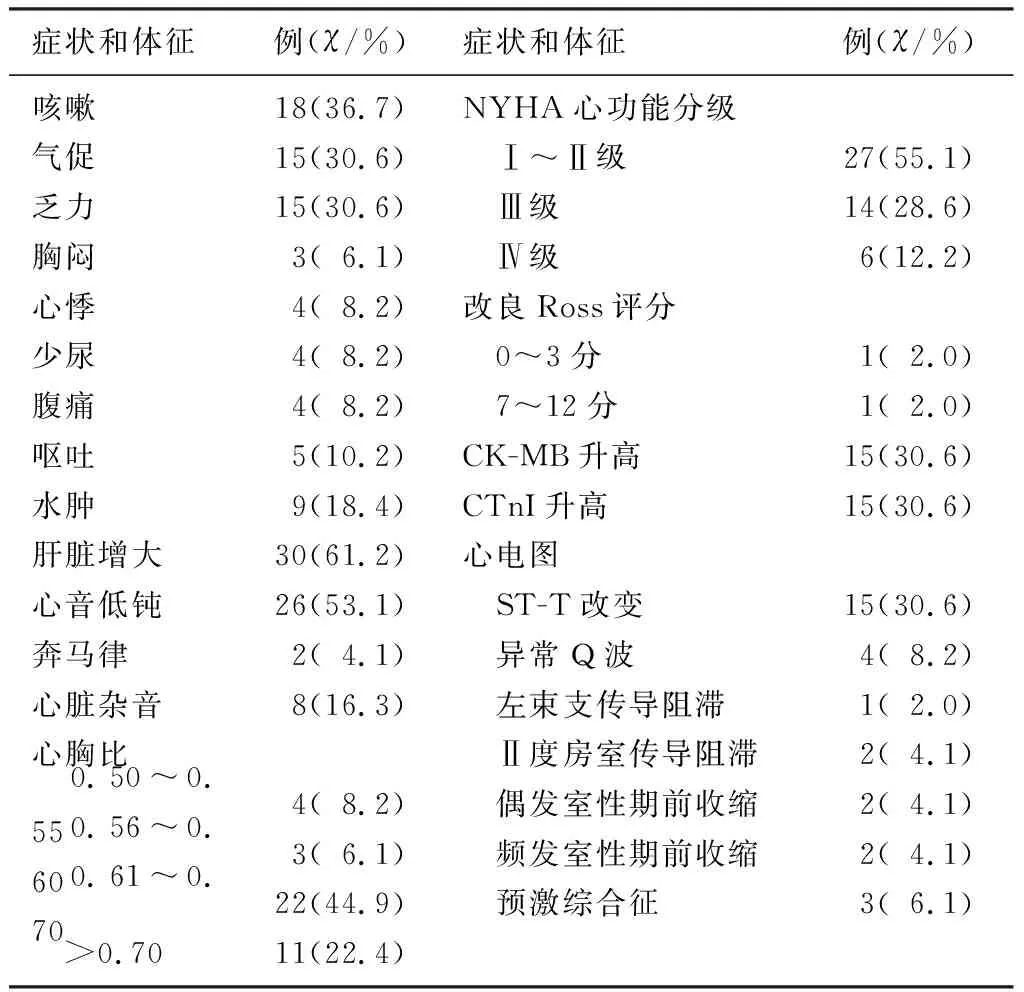
表1 DCM病兒的臨床基線資料

圖1 DCM病兒的Kaplan-Meier生存曲線
2.4 1例家族性DCM基因檢測結果
先證者,男,12歲,表現為嘔吐、乏力、尿少,體檢心律不齊、心音低鈍,肝臟肋下觸及2 cm。X線胸片示心胸比為0.7,肺炎改變。24 h動態心電圖示:竇性心律不齊;頻發室性期前收縮,共7 226次,多源,部分成對,呈聯律、間位,占總心搏的5.4%;交界性期前收縮,不完全性干擾性房室脫節;間歇性ST段改變;心率變異性中度降低。胸部B超檢查示雙側胸腔積液。心臟彩超檢查示:左房室明顯增大并室壁整體運動彌漫性減弱,左心室近心尖部豐富肌小梁回聲,二尖瓣與三尖瓣輕度反流;LVDd 64 mm,LVDs 57 mm,IVSd 7 mm,LVPwd 7 mm,LVEF 23%,LVFS 11%。發病后10個月因心力衰竭死亡;其姐于10歲時在外院診斷為DCM,診斷后數月因心力衰竭死亡。其三代家系圖譜見圖2。
先證者的家庭成員的基因突變位點見表2,先證者的家庭成員均無臨床癥狀及體征。先證者的DMD 基因出現 c.2473T>G的核苷酸變異,該變異導致了第825號的氨基酸由Trp變為Gly(p.Trp-825Gly);先證者TTN基因出現c.32186C>G的雜合核苷酸變異,該變異導致了第10729號的氨基酸由Thr變為Arg(p.Thr10729Arg);先證者SCN5A基因出現了c.2663T>C的雜合核苷酸變異,該變異導致了第888號的氨基酸由Phe變為Ser(p.Phe888Ser);先證者RBM20基因出現c.3512C>T的雜合核苷酸變異,該變異導致了第1171號氨基酸由Thr變為Met(p.Thr1171Met)。見圖3。先證者上述基因的突變均為錯義突變,非多態性變化。

:男性先證者;:女性死亡者。
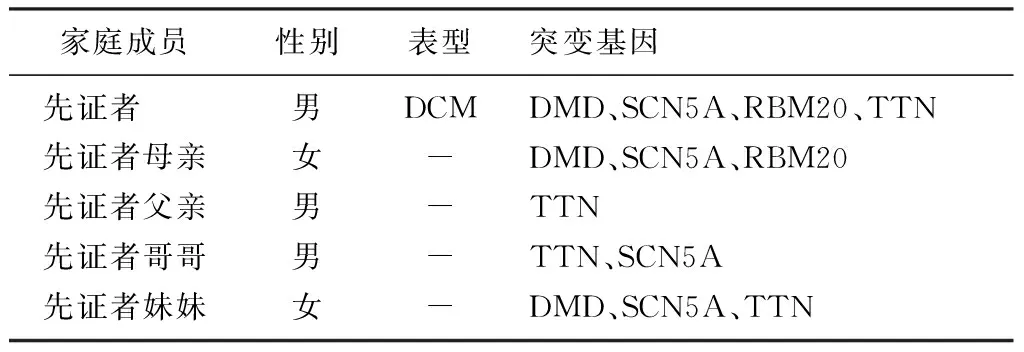
家庭成員性別表型突變基因先證者 男DCMDMD、SCN5A、RBM20、TTN先證者母親女-DMD、SCN5A、RBM20先證者父親男-TTN先證者哥哥男-TTN、SCN5A先證者妹妹女-DMD、SCN5A、TTN
3 討 論
DCM病兒發病年齡各家報道不一。美國多中心調查顯示,≤1歲嬰兒DCM的發病率明顯高于1歲以上的兒童[8],國內韓燕燕等[9]對62例DCM病兒的臨床資料進行分析發現,診斷年齡4.0~6.9歲者15例,7~14歲者34例,年長兒占79%;但是章旭等[10]的研究結果顯示,≤1歲的DCM病兒發病率最高,段慶寧等[11]的研究亦顯示DCM以≤2歲的兒童發病為主。分析發病年齡不一的原因可能與各研究的納入標準、樣本量及地域等因素有關。本組中DCM病兒<1歲者4例,1~5歲者13例,>5歲者32例(65.3%),提示廣西地區DCM的發病可能以年長兒居多。LIPSHULTZ等[8]的美國多中心兒童心肌病研究結果顯示,DCM的男女比例為1.5∶1,年發病率男孩高于女孩;NUGENT等[12]調查結果顯示,澳大利亞兒童心肌病男女比例為1.12∶1。國內也有小樣本資料顯示DCM病兒男性多于女性。本組DCM病兒中女性多于男性,與文獻報道結果不一致,推測其原因可能與樣本量小、廣西地區差異、性激素影響及X染色體突變等有關。

圖3先證者的基因突變
DCM病兒就診時多表現為心功能不全,少部分病兒出現嚴重心力衰竭;常合并呼吸道感染,甚至肺炎;部分病兒就診時已有CK-MB或者CTnI升高。DCM病兒心電圖以ST-T段改變、異常Q波、室性期前收縮等異常為主,超聲心動圖常提示心腔擴大,收縮功能減弱,少部分病兒合并心腔內附壁血栓等。DCM的臨床診斷并不困難,超聲心動圖能直接觀察心臟形態學和血流動力學的變化,具有準確、便捷、無創、重復性好、可動態觀察、無電離輻射等優點,是診斷DCM的首選方法[13]。
DCM預后的相關危險因素目前尚無統一標準,有研究表明發病年齡>5歲是兒童DCM的獨立危險因素[11],但ALVAREZ等[14]則認為發病年齡>6歲、合并先天性心臟病以及起病時LVEF低是兒童DCM的獨立危險因素。目前文獻報道的DCM生存率差異較大。國內有研究顯示DCM病兒的5年生存率為44.79%~48.00%[10,15];TSIRKA等[16]隨訪了91例DCM的病兒,其5年的生存率為83%;TOWBIN等[4]對美國和加拿大1 426例DCM病兒進行了系統研究,其5年病死率與心臟移植發生率為46%。本組DCM病兒的1、3和5年生存率分別為68.2%、54.4%和54.4%,其診斷后2年內病死風險高。但由于本組DCM病兒的失訪率較高,因此其精確的長期生存率仍需進一步評估。但規范的藥物治療能改善DCM病兒的癥狀,提高生活質量。
DCM的病因復雜,最常見的是心肌炎[17-19]。心肌炎引起DCM的病理生理機制涉及免疫炎癥反應導致的心肌纖維化、心室重構、心力衰竭、心肌局部微環境的改變、膠原合成和分解動態平衡間的互相作用等。除強心、利尿、ACEI類等抗心力衰竭、抗心律失常治療外,炎癥性DCM免疫抑制治療也具有一定的療效[20]。YOSHIKAWA等[21]證實了使用新型色氨酸柱的免疫吸附治療能改善由DCM引起的難治性心力衰竭病人的癥狀和運動耐受能力,在具有高自身抗體評分的病人中特別有效。炎癥性DCM的預后較家族遺傳性DCM的預后好[4]。本研究中2例家族遺傳性DCM病兒發病后病情進展快,均預后不良。因而明確病因對治療及判斷預后有重要價值。
兒童DCM中超過40%的病兒具有家族遺傳傾向[22],一個家系中包括先證者在內有≥2例DCM病人可診斷為家族性DCM[23]。其主要遺傳方式為常染色體顯性遺傳,其次為性連鎖遺傳、常染色體隱性遺傳及線粒體遺傳。目前已發現超過40個不同的基因突變可導致DCM[24-26]。杜興氏肌營養不良基因(DMD)是人類最大的基因,其突變可導致細胞骨架結構的異常[27],主要引起3種疾病,即DUCHENNE型肌營養不良(DMD)、BECKER型肌營養不良(BMD)以及X連鎖家族性DCM(XLFDCM)。XLFDCM是一種主要侵犯年輕男性的DCM,常呈快速進展的充血性心力衰竭,一般不出現骨骼肌疾病的臨床癥狀[28-30]。HOOGERWAARD等[31]在研究中發現,DMD以及BMD家系中的女性攜帶者大約22%出現DCM,其機制可能是X染色體失活;劉劍等[28]在X-連鎖DCM病人DMD基因突變分析及臨床評價中發現,4例女性攜帶者中1例為DCM,1例呈早期心肌病的表現,提示DMD基因突變所致XLFDCM家系中的女性攜帶者也有患病的可能。本組先證者DMD c.2473T>G突變導致第825號氨基酸由Trp變為Gly(p.Trp825Gly)的錯義突變,遺傳來源于先證者母親,先證者母親為雜合子,先證者為半合子,符合X連鎖隱性遺傳方式;先證者母親是雜合子,至今無心肌病表現。先證者姐姐10歲時因DCM死亡,雖缺乏其基因測定結果,但我們推測病兒的姐姐可能是雜合子,突變來自于先證者母親,可能因X染色體失活而發病;先證者妹妹也攜帶該基因,有DCM的發病風險,建議對其進行長期的隨訪觀察。先證者家系的特點符合XLFDCM,從家系基因檢測結果中可以推斷,DMD c.2473T>G(p.Trp825Gly)突變可能是導致先證者發病的致病突變,但需進一步行全面的家系基因篩查及對照研究加以驗證。
應該值得注意的是,本研究中本例先證者檢測到的TTN c.32186C>G(p.Thr10729Arg)、SCN5A c.2663T>C(p.Phe888Ser)以及RBM20 c.3512C>T(p.Thr1171Met)的雜合錯義突變,雖均為非多態性變化,但其致病性尚未見文獻報道;鑒于絕大多數心肌病是單基因病[32],因而這些基因突變在本家系中的作用有待進一步研究。隨著遺傳學技術的進展及二代基因測序的出現,為DCM的精準診斷提供了強有力的工具,如何對測序數據進行分析,識別出真正的致病突變成為新的挑戰[33-34]。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
少兒美術·書法版(2021年11期)2021-10-20 06:23:28
少兒美術·書法版(2021年8期)2021-10-20 06:08:10
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
雜文選刊(2016年7期)2016-08-02 08:39:56
小天使·一年級語數英綜合(2016年6期)2016-05-14 12:21:05
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
河南醫學研究(2014年5期)2014-02-27 14:52:41
