載亞硒酸鈉殼聚糖微球的制備、表征及緩釋性能研究
2018-10-10 10:45:44馳商龍臣吳少魏李光大劉信平
食品與機(jī)械 2018年8期
關(guān)鍵詞:殼聚糖
張 馳商龍臣吳少魏李光大劉信平
(1. 湖北民族學(xué)院生物科學(xué)與技術(shù)學(xué)院,湖北 恩施 445000;2. 湖北民族學(xué)院化學(xué)與環(huán)境工程學(xué)院,湖北 恩施 445000;3. 河南科技大學(xué)醫(yī)學(xué)技術(shù)與工程學(xué)院,河南 洛陽(yáng) 471003)
近年來(lái),被譽(yù)為“長(zhǎng)壽元素”和“抗癌之王”的人體生命活動(dòng)必需營(yíng)養(yǎng)微量元素硒[1-3],因世界許多地區(qū)(中國(guó)72%的地區(qū))嚴(yán)重缺乏[4],使得全民補(bǔ)硒熱潮在國(guó)內(nèi)外悄然興起。目前補(bǔ)硒方式主要有食補(bǔ)、攝入富硒保健品和藥補(bǔ)[5],補(bǔ)硒模式受價(jià)格昂貴和盲目進(jìn)補(bǔ)兩大瓶頸因素的制約。特別是盲目進(jìn)補(bǔ),硒與其他人體必需微量元素一樣具有兩重性[6-7],適量有益,超量中毒有害,成人每日攝人硒量高達(dá)400~800 mg/kg·體重可導(dǎo)致急性中毒,每天攝入2 400~3 000 μm 硒數(shù)月即出現(xiàn)慢性中毒癥狀。亞硒酸鈉已作為重要的補(bǔ)硒物被列為食品添加劑,如何把控補(bǔ)硒量的尺度,實(shí)現(xiàn)科學(xué)補(bǔ)硒成為關(guān)鍵。
天然高分子陽(yáng)離子多糖——?dú)ぞ厶牵云淞己玫奈叫浴⒊赡ば院蜕锵嗳菪裕挥米魉幬锞忈屳d體材料的研究已有相關(guān)報(bào)道[8-10],但可控緩釋硒材料的研究國(guó)內(nèi)外尚少見(jiàn)報(bào)道,文獻(xiàn)[11]報(bào)道將單質(zhì)硒或亞硒酸離子直接涂層成鍍硒鈦材料,用作載硒骨修復(fù)材料,但該硒的負(fù)載方式存在突釋效應(yīng),短時(shí)間易引發(fā)硒中毒。本研究以殼聚糖為壁材,采用乳化交聯(lián)法[12-13]制備殼聚糖載硒微球,讓硒長(zhǎng)時(shí)間緩慢釋放有效作用濃度,實(shí)現(xiàn)硒劑量的控制,以期開(kāi)辟科學(xué)補(bǔ)硒的新途徑。
1 材料與方法
1.1 材料與試劑
殼聚糖(Chitosan,CS):脫乙酰度>90%,上海伯奧生物科技有限公司;
亞硒酸鈉、乙酸、鹽酸、戊二醛、液體石蠟、Span-80、石油醚、丙酮、無(wú)水乙醇、氯化鈉、碳酸氫鈉、氯化鉀、三水合磷酸氫二鉀、六水合氯化鎂、氯化鈣、硫酸鈉、三羥甲基氨基甲烷等:分析純,上海國(guó)藥集團(tuán)化學(xué)試劑有限公司;
硒標(biāo)準(zhǔn)溶液:100 μg/mL,中國(guó)計(jì)量科學(xué)研究院。
1.2 儀器與設(shè)備
低速臺(tái)式離心機(jī):TDL-80-2B型,上海安亭科學(xué)儀器廠;
pH計(jì):PHSJ-3F型,上海精密科學(xué)儀器有限公司;
移液槍?zhuān)篗icroPette型,Dragon Laboratory Instruments Limited;
傅里葉紅外光譜儀:Nicolet iS10型,美國(guó)ThermoFisher Scientific公司;
微波消解儀:MARS HACKER 型,美國(guó)CEM Corporation公司;
雙道原子熒光光度計(jì):AFS-9760型,北京海光儀器公司;
SEM掃描電鏡:JSM-7001F型,日本電子株式會(huì)社。
1.3 方法
1.3.1 載硒、空白CS微球的制備
(1) 水相制備:稱(chēng)取一定量的殼聚糖粉末于體積分?jǐn)?shù)2%的醋酸溶液中,磁力攪拌使殼聚糖完全溶解,得質(zhì)量濃度2%的殼聚糖醋酸溶液,靜置過(guò)夜使其澄清透明,此為制備空白殼聚糖微球所需水相;準(zhǔn)確稱(chēng)取一定質(zhì)量的亞硒酸鈉于體積分?jǐn)?shù)2%的醋酸溶液中,配制成不同質(zhì)量分?jǐn)?shù)的含硒醋酸溶液,再將一定質(zhì)量的殼聚糖粉末置于此溶液中,磁力攪拌使殼聚糖完全溶解,配制成含硒量不同的殼聚糖溶液,靜置過(guò)夜使其澄清透明,此為制備載硒微球所需的水相。
(2) 油相制備:量取一定量的Span-80于液體石蠟中,攪拌均勻,配制成含5 mL/100 mL Span-80的石蠟油體系,此為制備微球所需油相。
(3) 載硒、空白CS微球的制備:在一定溫度條件下(空白微球35 ℃條件下),按水∶油體積比1∶5將水相緩慢滴加到油相中,邊滴加邊攪拌,待水相滴加完全后,繼續(xù)攪拌1 h,使水油兩相充分乳化,緩慢滴加一定量的交聯(lián)劑(12.5%的戊二醛),繼續(xù)攪拌1 h,使戊二醛與殼聚糖充分交聯(lián),靜置分層,去除上層液相,收集沉淀物,用石油醚和無(wú)水乙醇分別抽濾洗滌數(shù)次直至濾液澄清透明,此即為載硒、空白殼聚糖微球。滴加數(shù)滴丙酮,使微球顆粒相互分散,于45 ℃烘箱中烘干,得載硒和空白殼聚糖微球成品。
1.3.2 載硒CS微球制備的單因素試驗(yàn) 固定交聯(lián)劑用量為5%,反應(yīng)溫度35 ℃,投入Na2SeO3量為0.2%,乳化時(shí)間1 h,交聯(lián)時(shí)間1 h,分別在殼聚糖濃度為0.5%,1.0%,2.0%,3.0% 的條件下進(jìn)行試驗(yàn),考察殼聚糖濃度對(duì)微球載藥量、包封率[11]的影響;固定交聯(lián)劑用量為5%,反應(yīng)溫度35 ℃,殼聚糖濃度為2%,乳化時(shí)間1 h,交聯(lián)時(shí)間1 h,分別在Na2SeO3質(zhì)量濃度為0.2%,0.4%,0.8%,1.6%的條件下進(jìn)行試驗(yàn),考察Na2SeO3濃度對(duì)微球載藥量、包封率的影響;固定交聯(lián)劑用量5%,Na2SeO3質(zhì)量濃度為0.2%,殼聚糖濃度為2%,乳化時(shí)間1 h,交聯(lián)時(shí)間1 h,改變反應(yīng)溫度為35,45,55,65 ℃的條件下進(jìn)行試驗(yàn),考察溫度對(duì)微球載藥量、包封率的影響;固定反應(yīng)溫度35 ℃,Na2SeO3質(zhì)量濃度為0.2%,殼聚糖濃度2%,乳化時(shí)間1 h,交聯(lián)時(shí)間1 h,分別在交聯(lián)劑用量為2.5%,5.0%,10.0%,15.0%的條件下進(jìn)行試驗(yàn),考察交聯(lián)劑用量對(duì)微球載藥量、包封率的影響。微球的載硒量、包封率按式(1)、(2)計(jì)算:
(1)
(2)
式中:
c——微球的載硒量,%;
d——微球的包封率,%;
m1——微球的質(zhì)量,g;
m2——微球中硒的含量,g;
m3——投入微球中的硒質(zhì)量,g。
1.3.3 載硒殼CS微球制備的正交試驗(yàn) 根據(jù)單因素試驗(yàn)結(jié)果,選擇Na2SeO3濃度、殼聚糖濃度、反應(yīng)溫度及交聯(lián)劑用量4個(gè)因素為自變量,包封率和載硒量為因變量,設(shè)計(jì)L9(34)正交試驗(yàn)優(yōu)化微球制備工藝。
1.3.4 載硒CS微球體外緩釋試驗(yàn) 參照文獻(xiàn)[14~15]方法在聚乙烯燒杯中配制pH為7.40的模擬體液(SBF)。準(zhǔn)確稱(chēng)取0.04 g含硒微球于10 mL聚乙烯樣品管中,并向管中加入10 mL模擬體液(SBF),置于37 ℃恒溫水浴鍋中,定期從各管中取緩釋液且采用原子熒光光譜法測(cè)硒含量[14],并立即向管中添加緩釋液保證緩釋體系體積恒定不變。
2 結(jié)果與分析
2.1 單因素對(duì)載硒CS微球制備的影響
2.1.1 殼聚糖濃度對(duì)載硒CS微球制備的影響 殼聚糖濃度對(duì)載硒CS微球制備的影響見(jiàn)圖1。由圖1可知,殼聚糖濃度在0.5%~3.0%內(nèi),隨著濃度的升高,微球包封率與之呈正比關(guān)系,而載硒量卻呈負(fù)相關(guān)關(guān)系,隨之降低,而0.5%的殼聚糖溶液制備的微球壁薄不利于硒緩釋?zhuān)噬釛壴撟兞克阶髡辉囼?yàn)。
2.1.2 Na2SeO3濃度對(duì)載硒CS微球制備的影響 Na2SeO3濃度對(duì)載硒CS微球制備的影響見(jiàn)圖2。由圖2可知,在Na2SoO2質(zhì)量濃度為0.2%~1.6%時(shí),隨著Na2SeO3濃度的升高,微球的包封率隨之降低,載藥量隨之升高,Na2SeO3濃度為1.6%時(shí)微球包封率相對(duì)太低,故正交試驗(yàn)變量選0.2%,0.4%,0.8%三水平。
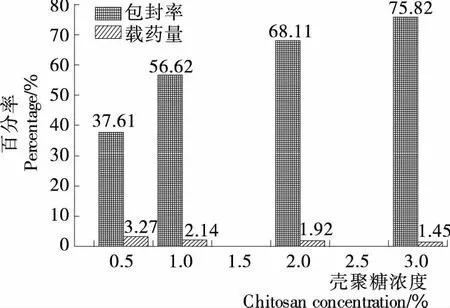
圖1 殼聚糖濃度對(duì)微球包封率和載藥量的影響Figure 1 Effects of CS concentration on encapsulation Efficiency and selenium loading capacity
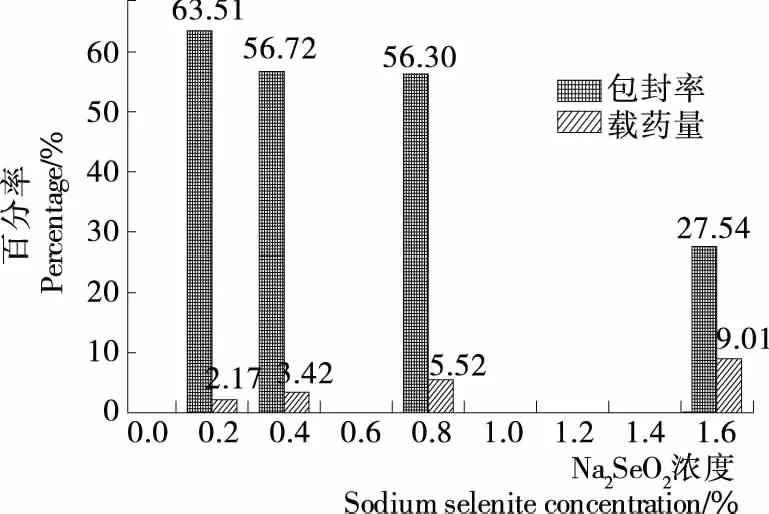
圖2 亞硒酸鈉濃度對(duì)微球包封率和載藥量的影響Figure 2 Effects of Na2SeO3 concentration on encapsulation Efficiency and selenium loading capacity
2.1.3 交聯(lián)溫度對(duì)載硒CS微球制備的影響 溫度對(duì)載硒CS微球制備的影響見(jiàn)圖3。由圖3可知,在溫度為35~65 ℃ 時(shí),不同溫度對(duì)微球得率略有影響,55 ℃時(shí)微球的包封率較好,而微球載藥量的峰值則出現(xiàn)在45 ℃,35 ℃時(shí)微球不僅包封率較低,且微球間黏連較多,正交試驗(yàn)選擇45,55,65 ℃三溫度水平。
2.1.4 交聯(lián)劑用量對(duì)載硒CS微球制備的影響 交聯(lián)劑用量對(duì)載硒CS微球制備的影響如圖4所示,由圖4可知,在交聯(lián)劑用量為2.5%~15%時(shí),隨著交聯(lián)劑用量的增加,微球的包封率和載藥量都呈先升高后降低的趨勢(shì),當(dāng)交聯(lián)劑的用量為水相用量的10.0%時(shí),微球包封率都達(dá)到最大值。當(dāng)交聯(lián)劑的用量為水相用量的5.0%時(shí),微球的載藥量達(dá)到最大值,綜合考慮,正交試驗(yàn)交聯(lián)劑用量選5%,10%,15%三水平。
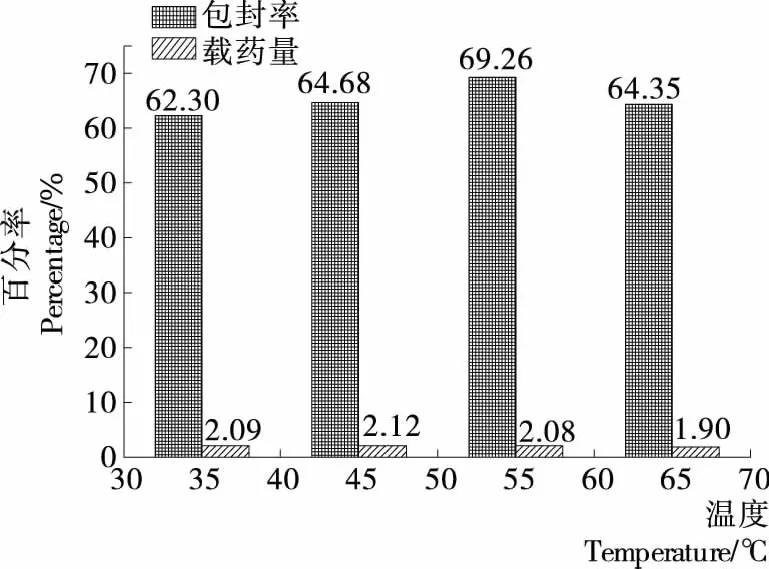
圖3 溫度對(duì)微球包封率和載藥量的影響
圖3 Effects of different temperature on encapsulation efficiency and selenium loading capacity
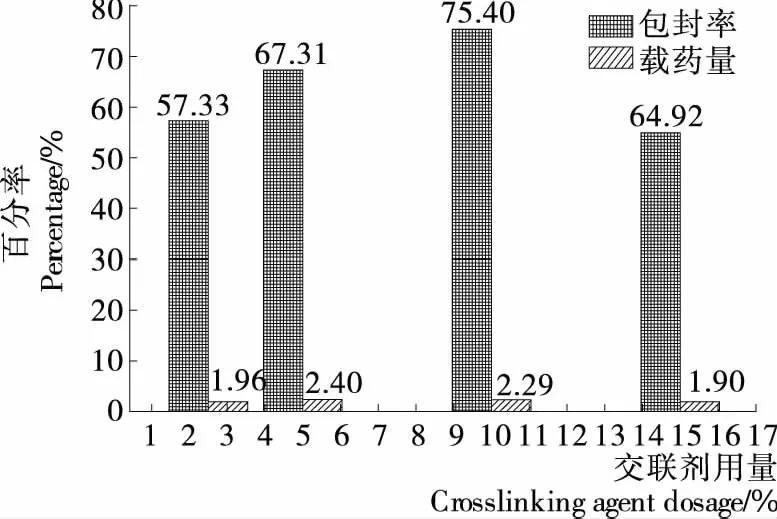
圖4 交聯(lián)劑用量對(duì)微球包封率和載藥量的影響
圖4 Effects of crosslinking agent on encapsulation efficiency and selenium loading capacity
2.2 正交試驗(yàn)
正交試驗(yàn)方案及采用SPSS 20.0軟件統(tǒng)計(jì)分析的試驗(yàn)結(jié)果見(jiàn)表1和表2。
2.2.1 包封率為因變量的正交試驗(yàn) 以包封率為因變量的正交試驗(yàn)結(jié)果方差分析見(jiàn)表3。
由表3可知,因素A、B、C、D對(duì)微球的包封率有顯著的影響(P<0.01)。根據(jù)表2、3結(jié)合變量因子對(duì)微球包封率影響的Duncan多重分析,得到微球最佳的優(yōu)化組合為A3B3C3D2。
2.2.2 載硒量為因變量的正交試驗(yàn) 以包封率為因變量的正交試驗(yàn)結(jié)果方差分析見(jiàn)表4。
由表4可知,因素A、B、C、D對(duì)微球的包封率有顯著的影響(P<0.01),根據(jù)表2、4以及變量因子對(duì)微球載硒量影響的Duncan多重分析,可知微球最佳的優(yōu)化組合為A1B3C3D2。
由上述可知,以不同的因變量進(jìn)行正交分析得到了不同的優(yōu)化組合條件,分別為A3B3C3D2、A1B3C3D2。按照這些組合條件設(shè)計(jì)進(jìn)行驗(yàn)證實(shí)驗(yàn),結(jié)果見(jiàn)表5。
由表5可知,驗(yàn)證實(shí)驗(yàn)結(jié)果與正交分析結(jié)果相吻合。A1B3C3D2組正交優(yōu)化方案的載硒量更優(yōu)、A3B3C3D2組的包封率更好。
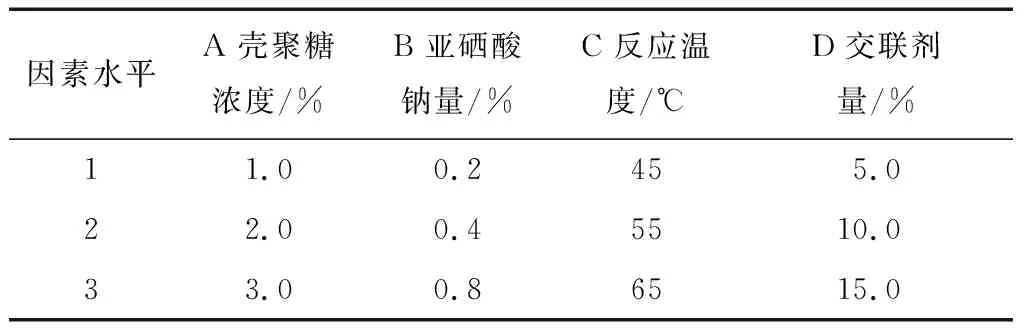
表1 乳化交聯(lián)法正交試驗(yàn)因素水平Table 1 Factor levels of orthogonal test with emulsion cross-linking method
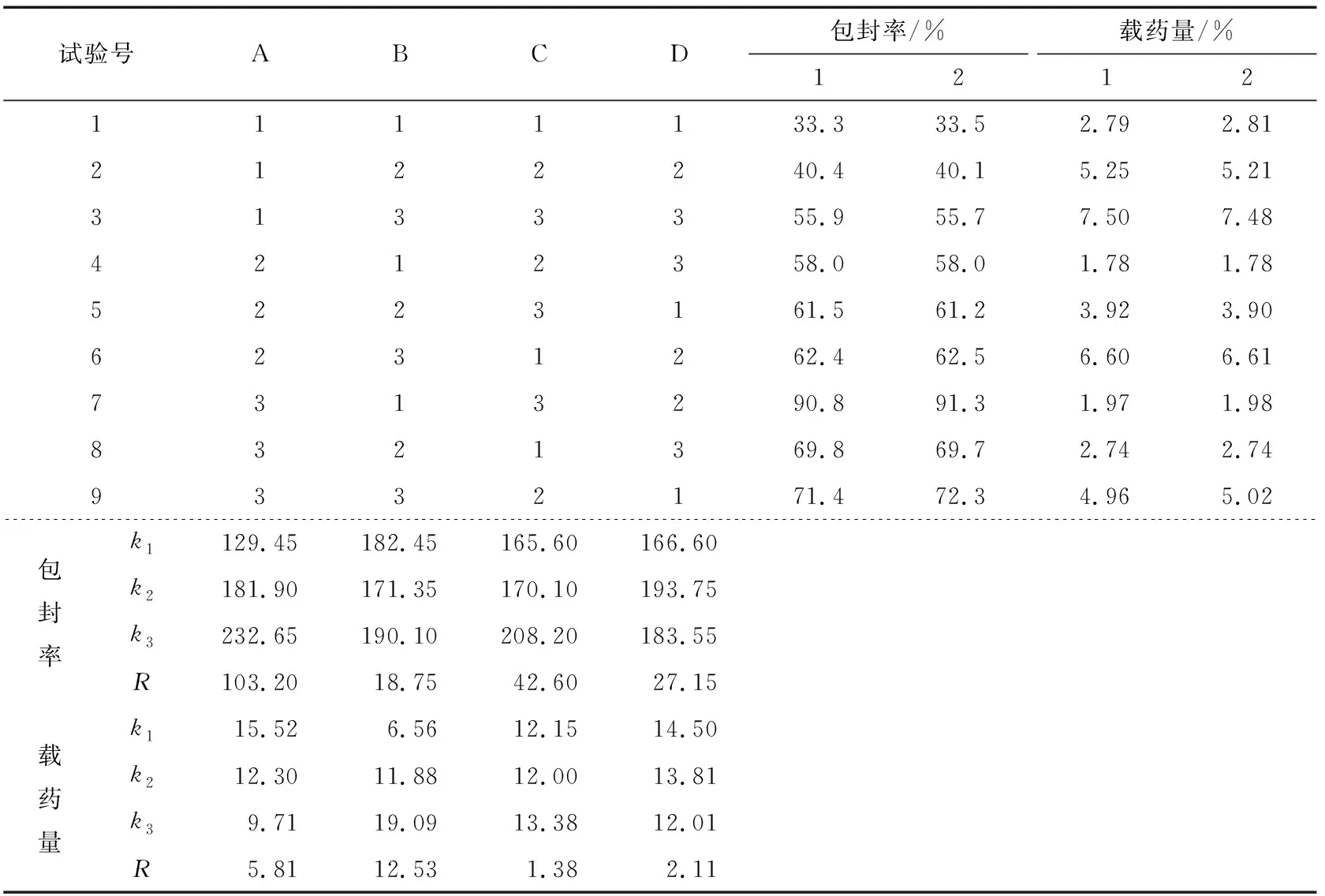
表2 L9(34)正交試驗(yàn)方案及結(jié)果Table 2 Scheme and results of orthogonal experimental scheme L9(34)
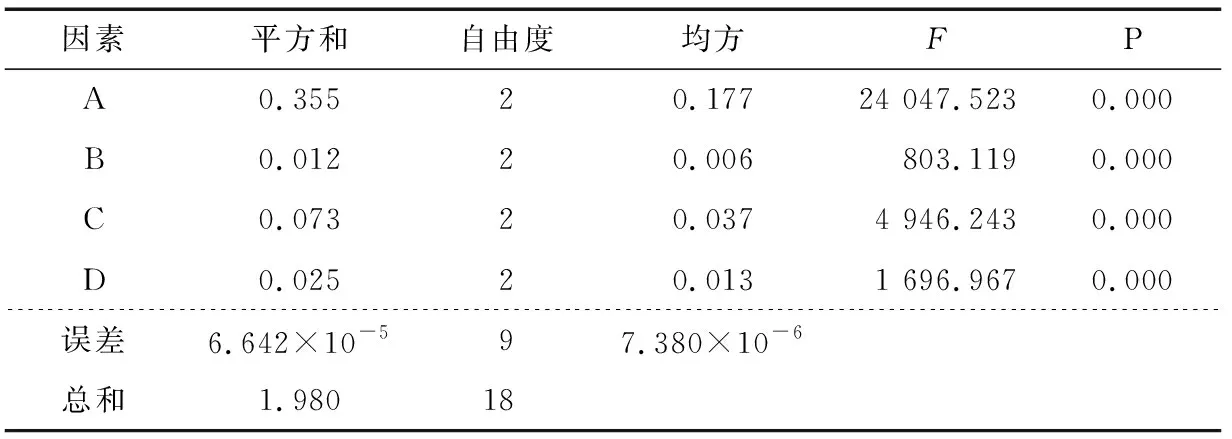
表3 正交試驗(yàn)結(jié)果的方差分析Table 3 Variance analysis of orthogonal test
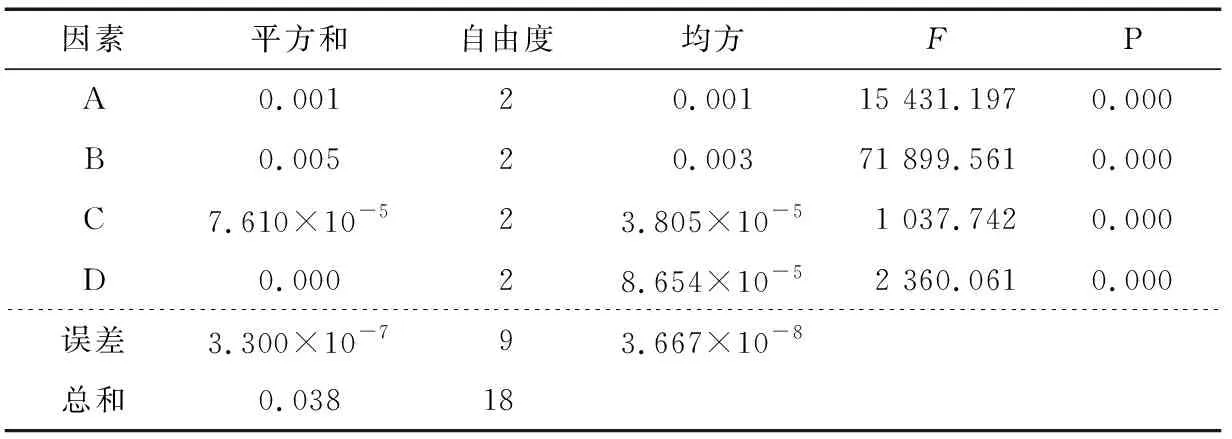
表4 正交試驗(yàn)結(jié)果的方差分析Table 4 Variance analysis of result on orthogonal test
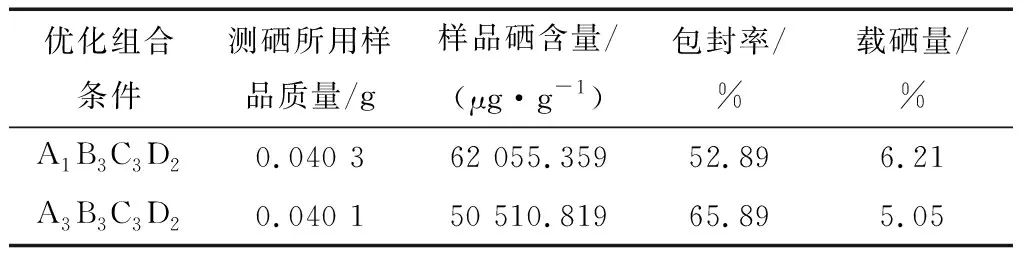
表5 驗(yàn)證實(shí)驗(yàn)結(jié)果Table 5 Result of test and verify experimentation
2.3 載硒CS微球的形貌表征
將采用優(yōu)化組合方案所制備的微球進(jìn)行SEM電鏡掃描觀察,微球的SEM圖片見(jiàn)圖5。從圖5可發(fā)現(xiàn),A1B3C3D2和A3B3C3D22方案條件下制備的微球都有較好的球形結(jié)構(gòu),形態(tài)良好,但A1B3C3D2條件下制備的微球粒徑(粒徑為4~8 μm)顯著小于A3B3C3D2(粒徑為9~12 μm),說(shuō)明隨著

a、c. A3B3C3D2方案的空白/載硒微球;b、d. A1B3C3D2方案的空白/載硒微球
圖5 載硒殼聚糖微球的SEM圖
Figure 5 SEM images for blank and selenium-loaded microspheres
材壁殼聚糖濃度的增大,所制備的微球粒徑也增大。
2.4 載硒CS微球的IR和熱分析
2種方案制得微球的IR和熱分析結(jié)果基本相同,紅外光譜、DTG和TG見(jiàn)圖6~8。
2.4.1 紅外分析 從圖6可知,殼聚糖的特征吸收峰在3 441.03 cm-1處出現(xiàn)的寬峰是O—H和N—H的伸縮振動(dòng)峰,2 908.98 cm-1為CH3的C—H伸縮振動(dòng)吸收峰,2 883.02 cm-1是醛基的特征峰,1 652.01,1 596.75 cm-1是酰胺鍵Ⅰ和酰胺鍵Ⅱ的特征吸收峰,1 381.03 cm-1附近是CH3的C—H的對(duì)稱(chēng)彎曲振動(dòng)峰,1 424.71 cm-1附近是—CH2的彎曲振動(dòng)吸收,1 325.76,1 264.26 cm-1是殼聚糖中C—N的伸縮振動(dòng)峰,1 079.74 cm-1附近是殼聚糖的C—OH 伸縮振動(dòng)吸收峰;空白殼聚糖微球的IR曲線與殼聚糖相比,在1 079.74 cm-1以下的低波數(shù)段曲線形狀相對(duì)于高波數(shù)段變化較大,3 441.03 cm-1處的峰基本未變,2 923.73 cm-1處較弱的吸收峰,是微球中尚存有少量未發(fā)生反應(yīng)的醛基,1 638.58 cm-1處有較強(qiáng)的吸收峰,這是戊二醛和殼聚糖的氨基反應(yīng)形成的schiff堿吸收峰,即戊二醛與殼聚糖發(fā)生了交聯(lián)反應(yīng);從Na2SeO3的IR光譜圖可知:在786.94 cm-1附近是亞硒酸根Se═O的伸縮振動(dòng)峰,1 113.82 cm-1附近是亞硒酸根O—Se—O的對(duì)稱(chēng)伸縮振動(dòng)峰,489.29 cm-1處是O—Se—O彎曲變角振動(dòng)峰;載硒微球和空白微球的IR光譜曲線圖整體上比較相近,但在2 923.73 cm-1處的吸收峰非常微弱,說(shuō)明載硒殼聚糖微球中未發(fā)生反應(yīng)的醛基很少,1 409.78 cm-1處有強(qiáng)的吸收峰,這是載硒微球中O—Se—O的伸縮振動(dòng)峰,證明微球中的確包裹了預(yù)期硒目標(biāo)藥物,相對(duì)于Na2SeO3純物質(zhì),這個(gè)峰向高波數(shù)發(fā)生了明顯的移動(dòng)。
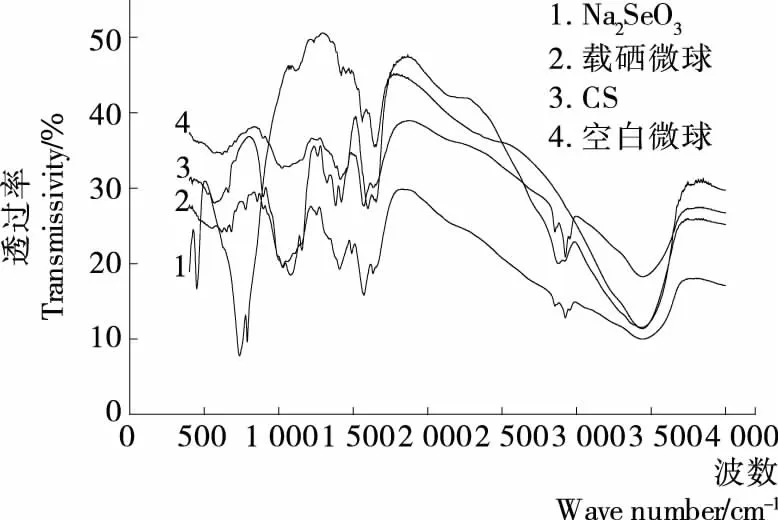
圖6 各樣品的紅外光譜曲線Figure 6 IR curves of samples
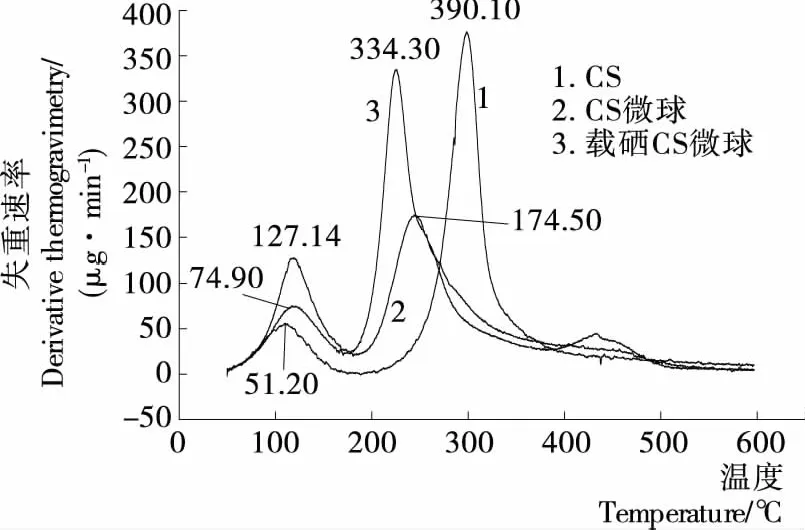
圖7 各樣品的DTG曲線Figure 7 DTG curves of samples
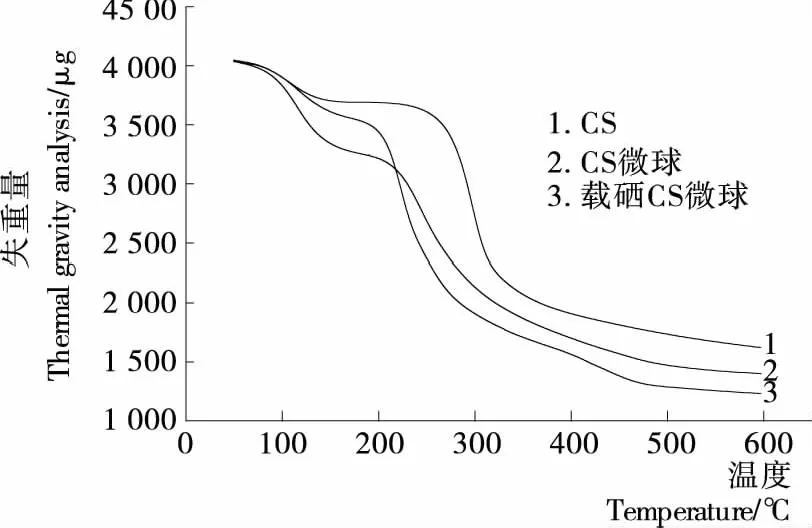
圖8 各樣品的TG曲線Figure 8 TG curves of samples
2.4.2 熱重分析 由圖7、8可知,總體上制備的微球熱穩(wěn)定性?xún)?yōu)于單純的殼聚糖。殼聚糖在溫度為67.24~133.31 ℃時(shí)有個(gè)小的失重階段,此時(shí)剩余質(zhì)量為樣品總重的99.32%~91.20%,102.60 ℃失重速率最大達(dá)到51.20 μg/min,此階段為自由水的揮發(fā);243.84~588.87 ℃時(shí)有較大的失重,剩余質(zhì)量為樣品總重的90.89%~40.07%,298.70 ℃失重速率最大達(dá)到390.10 μg/min,此為殼聚糖的部分分解。空白殼聚糖微球存在2個(gè)失重階段,69.14~161.79 ℃時(shí)剩余質(zhì)量為樣品總重的99.02%~84.34%,118.0 ℃失重速率最大達(dá)到127.14 μg/min;185.15~588.87 ℃時(shí)有較大的失重,剩余質(zhì)量為樣品總重的79.82%~32.71%,244.30 ℃ 失重速率最大達(dá)到174.50 μg/min。載硒殼聚糖微球存在2個(gè)失重階段,68.05~158.93 ℃時(shí),剩余質(zhì)量為樣品總重的99.48%~88.88%,117.8 ℃失重速率最大達(dá)到74.90 μg/min;177.54~588.38 ℃時(shí)有較大的失重,剩余質(zhì)量為樣品總重的87.71%~30.79%,225.50 ℃失重速率最大達(dá)到334.30 μg/min。
2.5 載硒CS微球體外緩釋性能的調(diào)控
將表1中9組正交試驗(yàn)組制備的載硒CS微球樣品進(jìn)行體外緩釋試驗(yàn),探求微球的緩釋可控性。準(zhǔn)確稱(chēng)取每1種待測(cè)樣品0.020 g 各3份,裝入透析袋中,將透析袋放入10 mL聚乙烯樣品管中,向其中加入10 mL模擬體液(SBF),37 ℃恒溫水浴,定期取出緩釋液5 mL檢測(cè)其硒含量,計(jì)算微球的累計(jì)釋放量,并同時(shí)補(bǔ)加同體積和溫度的SBF液。各樣品的緩釋曲線見(jiàn)圖9。圖9顯示,低濃度的CS和交聯(lián)劑用量少的試驗(yàn)組制備的微球,釋硒速率大于高濃度壁材和交聯(lián)劑用量多的試驗(yàn)組,試驗(yàn)組1的釋放速率最大且持續(xù)時(shí)間較短,在6 h內(nèi)累計(jì)釋放率達(dá)到59.6%,有一定的突釋現(xiàn)象,而3% CS和15%交聯(lián)劑的試驗(yàn)組制備的微球緩釋效應(yīng)明顯。即微球的釋硒速率受CS材壁濃度和交聯(lián)劑用量的影響很大,而交聯(lián)溫度和投藥量對(duì)釋放速率的影響相對(duì)弱化。由此可推斷,球壁厚度和交聯(lián)度是微球藥物釋放速率的主要控制因素,通過(guò)選擇相關(guān)變量因素的不同水平,調(diào)控球壁厚度和交聯(lián)度,可實(shí)現(xiàn)載硒微球硒的可控緩釋。
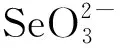
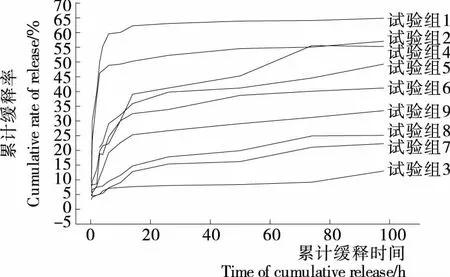
圖9 正交試驗(yàn)各組制備的Na2SeO3/CS微球緩釋曲線Figure 9 Sustained release selenium curve of Na2SeO3/CS microsphere on orthogonal test

圖10 載硒微球的體外累計(jì)釋放曲線Figure 10 Release curve of drug-loading microspheres in vitro
微球釋放介質(zhì)的pH變化并不明顯,僅在pH 7.3~7.4時(shí)有較小的波動(dòng),表明微球在釋硒過(guò)程中對(duì)釋放介質(zhì)的pH影響較小。
3 結(jié)論
本試驗(yàn)研究結(jié)果表明:載Na2SeO3殼聚糖緩釋微球最佳制備工藝條件為溫度65 ℃,殼聚糖濃度3%,亞硒酸鈉濃度0.8%,交聯(lián)劑用量15%,包封率和載藥量分別為65.89%,5.05%,且微球形貌優(yōu)良、平均粒徑為10 μm、熱穩(wěn)定性?xún)?yōu)于單純的殼聚糖。
體外緩釋試驗(yàn)證明,球壁厚度和交聯(lián)度是微球釋硒速率的主要控制因素,通過(guò)選擇相關(guān)變量因素的不同水平,調(diào)控這2個(gè)因素,可實(shí)現(xiàn)載硒微球硒的可控緩釋。最佳工藝制備的微球緩釋性能良好,緩釋速率在482 h后達(dá)平穩(wěn),有效緩釋時(shí)間達(dá)35 d,即載Na2SeO3殼聚糖微球具有較好的長(zhǎng)效緩釋能力。
因此乳化交聯(lián)法制備可控緩釋硒的殼聚糖微球是可行的,將該載硒殼聚糖微球應(yīng)用到食品、藥品及精細(xì)化工等行業(yè)產(chǎn)品中,能有效避免硒的突釋效應(yīng),為缺硒群體的科學(xué)補(bǔ)硒提供一條新途徑。后續(xù)試驗(yàn)需對(duì)該產(chǎn)品進(jìn)行體外細(xì)胞和體內(nèi)動(dòng)物試驗(yàn),以精準(zhǔn)評(píng)價(jià)其應(yīng)用過(guò)程中的生物安全性。
猜你喜歡
河北科技師范學(xué)院學(xué)報(bào)(2022年2期)2022-08-26 08:55:40
河北科技師范學(xué)院學(xué)報(bào)(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(shù)(2017年1期)2017-03-20 13:37:59
廣西科技大學(xué)學(xué)報(bào)(2016年1期)2016-06-22 13:10:38
天然產(chǎn)物研究與開(kāi)發(fā)(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國(guó)果菜(2015年2期)2015-03-11 20:01:01
應(yīng)用化工(2014年7期)2014-08-09 09:20:21
應(yīng)用技術(shù)學(xué)報(bào)(2014年4期)2014-02-28 14:52:40
