甲烷氧化偶聯制乙烯機理和動力學研究進展
2018-10-11 09:17:18張明森武潔花
石油化工 2018年9期
李 鵬,張明森,武潔花
(中國石化 北京化工研究院,北京 100013)
天然氣作為現代工業三大能源(煤、石油、天然氣)之一,具有優質、清潔、儲量豐富的優點。甲烷是天然氣中的主要成分,目前針對甲烷主要有兩個利用途徑[1]:一是甲烷的間接利用;二是甲烷的直接利用。甲烷的間接利用是甲烷通過水蒸氣重整反應生成的合成氣再進一步轉化成甲醇、合成氨、二甲醚等,這是甲烷最主要的利用途徑,但在甲烷間接轉化的兩個過程中都需要消耗大量的能量,因此甲烷的直接利用成為研究的重點。甲烷的直接轉化有多種途徑,可直接氧化偶聯生成乙烯和乙烷,也可選擇氧化生成甲醛和甲醇,還可直接部分氧化生成合成氣,由于產物中乙烯是石油生產中重要的化工基礎原料,甲烷的氧化偶聯生成乙烯成為研究的焦點。
1982年Keller等[2]提出甲烷氧化偶聯制乙烯的研究技術,并迅速引起了各國科學家的廣泛關注,但由于反應過程難以控制,研究成果一直未達到工業期望的效果。2006年,該研究方向伴隨納米技術和反應器設計理念的發展,再度呈現光明的前景[3]。催化反應動力學是研究催化反應速率與過程變量之間的關系。通過研究甲烷氧化偶聯反應機理和動力學,對反應催化劑的研發、反應器的研發和生產裝置實現最優化操作,具有重要的指導意義。
本文綜述了近年來甲烷氧化偶聯催化反應的主要反應機理模型和動力學模型,為催化劑和催化反應器的研究與設計提供依據。
1 甲烷氧化偶聯動力學反應機理
甲烷氧化偶聯反應的動力學模型與甲烷和O2結合的機理密切相關,不同的催化劑對應了不同的反應機理。關于甲烷氧化偶聯的反應機理,研究者們進行了大量的研究[4]。Morales等[5]通過 EPR 光譜發現了大量的甲基自由基和[LiO]+的存在,提出甲基自由基是該反應的中間產物。此后,研究者提出甲烷氧化偶聯反應分兩步走的反應機理:首先甲烷吸附在催化劑表面,脫去一個H原子生成甲基自由基,然后甲基自由基偶聯生成乙烷分子,乙烷分子脫氫生成乙烯。有文獻報道了甲烷氧化偶聯反應中O2分子的非解離吸附機制[6-7],也有文獻支持O2分子的解離吸附機制。Takanable等[8]進行了同位素干擾研究實驗(使用CD4/CH4,D2O/H2O,18O2/16O2),評估了Mn/Na2WO4/SiO2的動力學同位素效應,發現C—H鍵的激活是動力學不可逆的,他們還表明O2的解離是平衡的,但隨H2O/O2的質量比的增加,轉化率和停留時間增加,O2的這種平衡就會被打破,這也在一方面對O2的解離吸附提供了強有力的支持[9]。許多研究者根據不同的催化劑研究了甲烷氧化偶聯的反應機理[9-11]。Golpasha等[12]根據 Rideal-Redox 機理研究了 Na/Mg2O3的動力學,Iwamatsu等[13]研究了Rideal-Redox、Langmuir-Hinshelwood及Multiple-Redox機理,Miro等[14]分別根據 Rideal-Redox機理和Eley-Rideal機理研究了Li/NiTiO3和Na/NiTiO3的動力學模型。以下為幾個比較典型的反應機理模型
1.1 Mars-Van Krevelen 機理模型
Sohrabi等[15]用 CaTiO3催化劑,分別在 760,770,780 ℃溫度下進行實驗,結果發現當進料O2分壓較低時,實驗結果符合Mars-Van Krevelen機理。Mars-Van Krevelen機理認為O2首先在催化劑表面發生解離吸附,然后甲烷分子與催化劑表面的吸附氧結合。O2的吸附速率和甲烷的反應速率分別見式(1)和(2):

穩定狀態時,rO2= rCH4,則可得式(3):

1.2 Langmuir-Hinshelwood機理模型
Sohrabi等[15]在較高的 O2分壓下以 CaTiO3為催化劑進行實驗,發現實驗結果符合Langmuir-Hinshelwood機理。該機理認為催化劑表面的O2和甲烷分子的吸附是雙活性位點吸附,其中,O2屬于非解離吸附,在催化劑表面吸附O2分子和甲烷分子發生反應。穩定的條件下,O2的吸附和甲烷的吸附分別達到平衡。即有式(4):

進一步可得式(5):
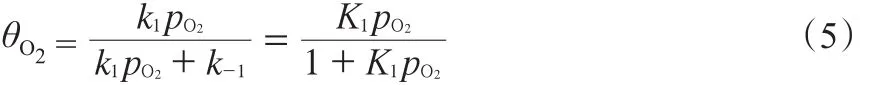
同理可求出被甲烷吸附的活性位點,見式(6):

甲烷的消耗速率見式(7):

由式(4)~(7)得式(8):
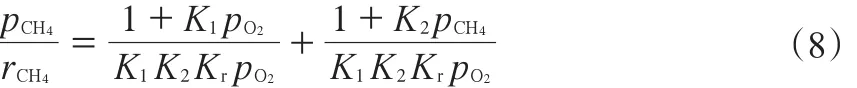
1.3 Eley-Rideal機理模型
Miro等[14]在一定條件下對加入1.6%(w)Na的鈦酸鎳催化劑進行研究,研究表明實驗結果與Eley-Rideal機理較符合。該機理認為O2首先在催化劑表面發生非解離吸附,然后吸附的O2與甲烷分子發生反應。穩定狀態下,O2的吸附和脫附達到平衡,甲烷的消耗速率見式(9):

根據式(5)進一步可得式(10):

1.4 Rideal-Redox機理模型
Seyed等[16]在不同的反應條件下,以Na/BaTiO3/MgO為催化劑進行實驗,實驗結果調查對比了Rideal-Redox的4個機理模型。
第一種模型是O2分子在催化劑表面進行可逆吸附,然后CH4氣體分子與吸附的O2進行反應生成CH3·,CH3·與O2分子反應生成CH3O2·,然后由CH3O2·轉化成COx,2個CH3·合成C2。在穩定的條件下,O2的吸附和脫附達到平衡,其中,O2脫附一部分參與甲烷的反應中。即有式(11):
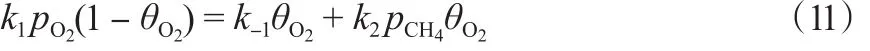
進一步可得式(12):

甲烷的反應速率見式(13):
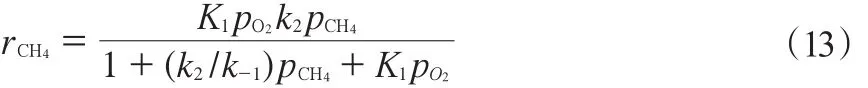
Seyed 等[16]還計算得出了 rC2H6,見式(14):

穩定條件下見式(15)和(16):
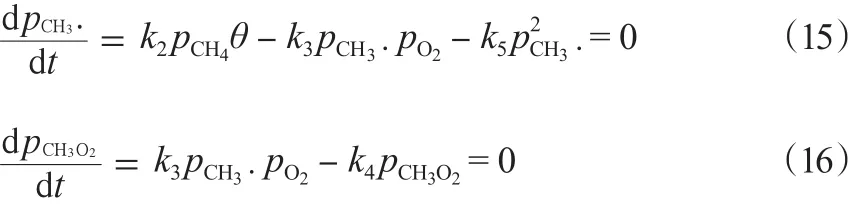
由見式(15)和(16)進一步可得rC2H6,見式(17):

該機理的第二種模型的第三步基元反應是可逆反應,第一步基元反應屬于非可逆反應。同第一種模型的計算方法,穩定條件下,O2的吸附與脫附達到平衡,O2的脫附全部參與甲烷的反應當中。可有式(18):

所以有式(19):
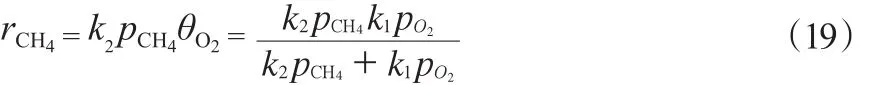
然后根據式(20)~(21)及式(14)可得到rC2H6。
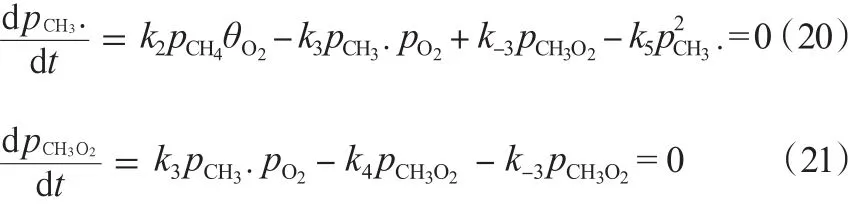
該機理的第三種模型主要考慮了O2的解離吸附。其中,,穩定條件下,O2的吸附與脫附達到平衡,O2的脫附全部參與甲烷的反應當中。由式(14)及式(22)~(25)可得rC2H6。
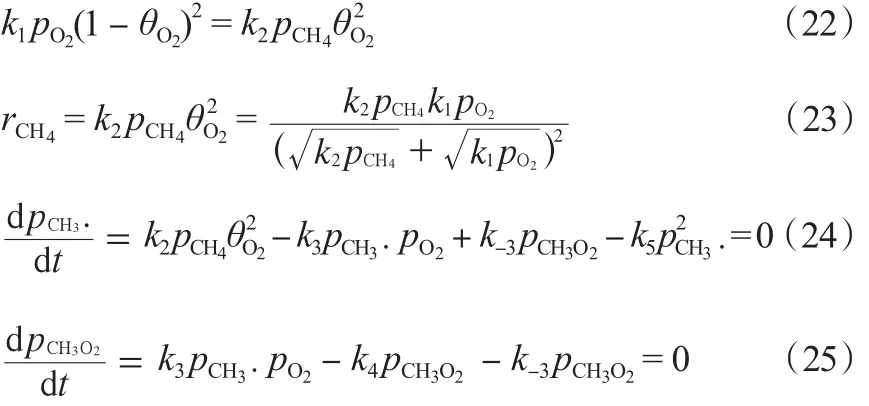
該機理的第四種模型指的是O2的解離吸附是一個可逆反應。穩定條件下,O2的吸附與脫附達到平衡,O2的脫附全部參與甲烷的反應當中。由式(14)及式(24)~(25)和式(26)~(27)可得 rC2H6:
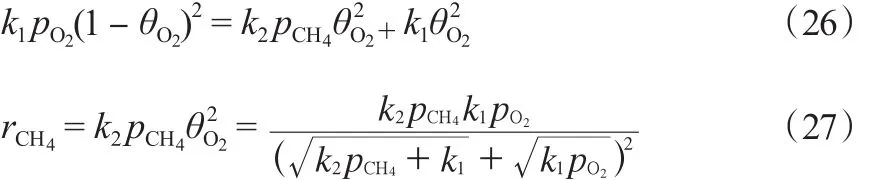
2 甲烷氧化偶聯反應動力學模型
研究反應動力學可以幫助人們了解催化劑的反應速率方程以及催化劑的詳細反應機理,對觀測到的反應數據進行解釋和優化,預測不同操作變量的影響,進而減少所需的實驗工作量,為催化反應器的設計提供依據和基礎。
對于甲烷氧化偶聯反應的動力學模型,研究者進行了大量的實驗研究[17-19]。Stansch 等[20]以反應機理和經驗相結合的方式總結了La2O3/CaO催化劑上的甲烷氧化偶聯的反應網絡和動力學模型,動力學方程中包括9個多相催化反應和1個均相催化反應;Vatania等[17]使用遺傳算法模擬了Li/MgO催化劑的動力學,提出了一個包含16個反應步驟的反應動力學模型,其中,包含3個主反應步驟和13個二級反應步驟;Sun等[21]提出Li/MgO催化劑和Sn/Li/MgO催化劑的一個動力學模型,其中,包括39個氣相基元步驟反應及14個催化反應,并比較了兩種催化劑的選擇性;Miro等[14]通過Na/NiTiO3催化劑和Li/NiTO3催化劑研究了甲烷氧化偶聯反應動力學,分別通過Rideal-Redox機理和Ely-Rideal機理得到了動力學研究結果;Wolf等[22]研究了CaO/CeO2催化劑的甲烷氧化偶聯反應動力學,通過動力學分析了催化劑和甲烷氧化偶聯反應的固相特性之間的關系;Robert等[23]研究了 La0.9Ce0.1CoO3催化劑的甲烷氧化偶聯動力學反應,其中,考慮了水和CO2對甲烷轉化率的作用,實驗結果表明,水對甲烷轉化率的影響更大。另外不同的學者使用不同的催化劑進行了甲烷氧化偶聯反應的反應器模擬。2001年,Tye等[24]為優化甲烷氧化偶聯制乙烯動力學模型,在不同的條件下使用Math CAD8.0軟件進行了反應器的一維模擬。2008年,Yaghobi等[25]使用代碼編寫了一個二維穩態反應器模型,通過三維數值模擬傳熱和非均勻動力學模型的流場耦合,研究了Sn/BaTiO3催化劑的甲烷氧化偶聯動力學。
由于不同的機理對應不同的模型,且甲烷氧化偶聯反應步驟較多,不同的學者針對不同的反應步驟也有不同的反應模型,研究學者根據不同的反應機理研究了甲烷氧化偶聯反應的動力學模型,主要的動力學模型為以下5種。
2.1 Nastaran Razmi Farooji模型
Farooji等[26]以 SnBaTiO3為催化劑,分別在725,750,775 ℃、烷氧摩爾比2.0~4.5、重時空速100 min-1的條件下研究了Mars-Van Krevelen 機理、Eley-Rideal機理、4種Rideal-Redox機理以及Langmuir-Hinshelwood機理,研究發現實驗結果和Langmuir-Hinshelwood機理比較符合,得出Nastaran Razmi Farooji動力學模型(見表1)。
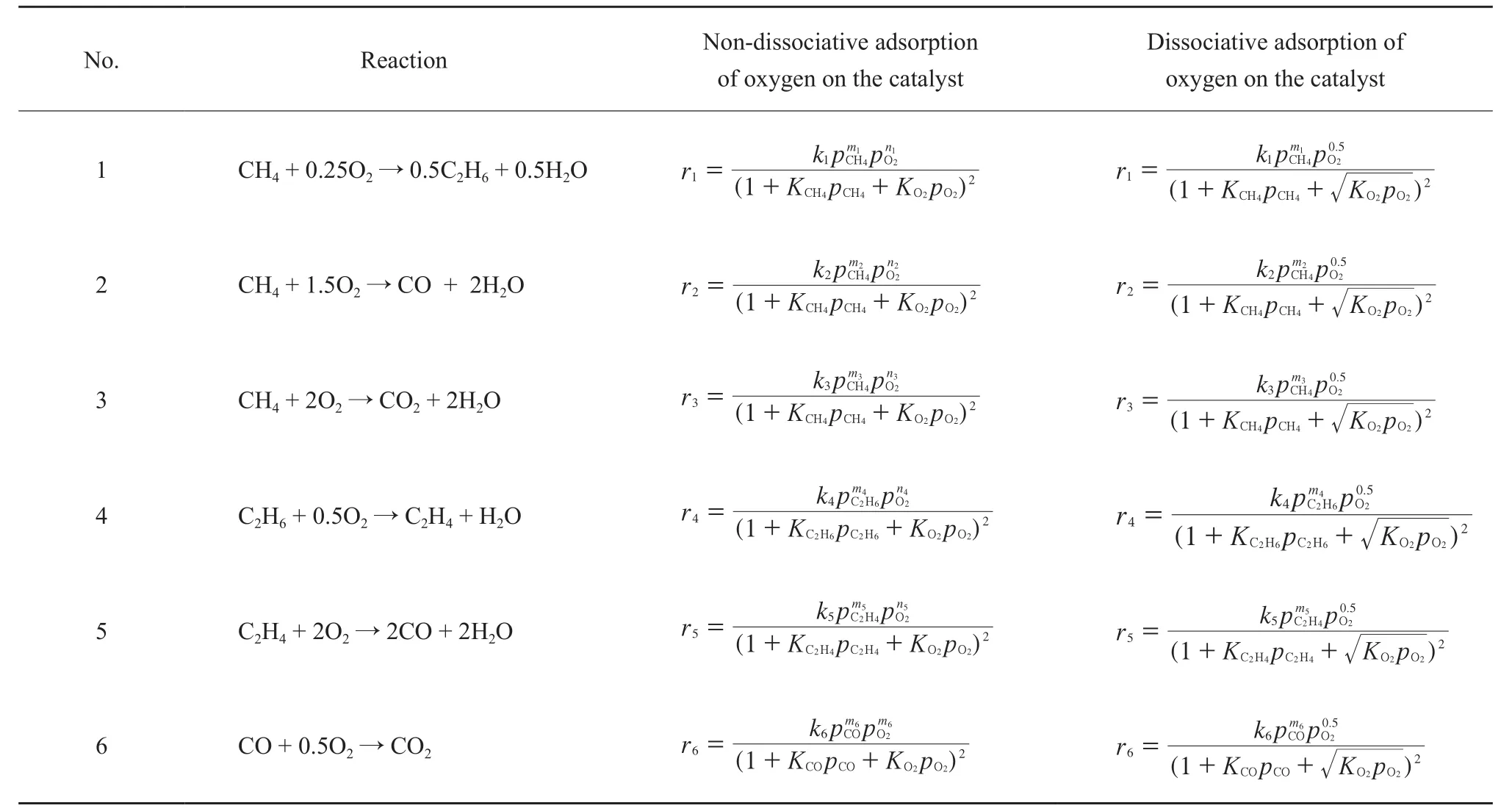
表 1 Nastaran Razmi Farooji模型[26]Table 1 Nastaran Razmi Farooji model
2.2 Sylvie Lacombe模型
Lacombe等[27]以 La2O3為催化劑,在 750 ℃下,控制甲烷的進料分壓為4.5~32 kPa、O2分壓為0.12~3.5 kPa、氣體進料的流動速率為0.33 cm3/s,測得出口各組分的分壓,得到的實驗數據與不同的動力學方程進行優化,實驗數據與Sylvie Lacombe模型較符合(表2)。
2.3 Stansch模型
Stansch等[20]以La2O3/CaO 為催化劑,在 O2分壓1~20 kPa,甲烷進料分壓10~95 kPa,溫度700~955 ℃的條件下進行實驗,實驗數據代入到Stansch模型的動力學方程中,實驗誤差均在20%以內。Stansch模型幾乎包括了其他文獻中用其他催化劑得出的所有反應步驟(表3),該模型也已經被不同的研究者使用[28-29]。Farsi等[30]對不同的動力學模型做了總結對比,指出Stansch機理模型相對其他模型,所得模擬結果與實驗結果更接近。Ghiasi等[31]也成功地將該模型應用于Na/W/Mn/SiO2催化劑上。
2.4 Kamali Shahri模型
以浸漬法制備的2%Mn/5%Na2WO4/SiO2為催化劑,在O2分壓4~20 kPa、甲烷進料分壓20~80 kPa、溫度800~900 ℃、O2沒有完全轉化的條件下進行實驗。Kamali Shahri反應動力學模型包含3個初級反應步驟和4個連續次級反應步驟(見表4),動力學模型符合Langmuir-Hinshelwood機理,實驗數據結果與動力學模型的模擬結果的誤差在 20% 以內[32]。
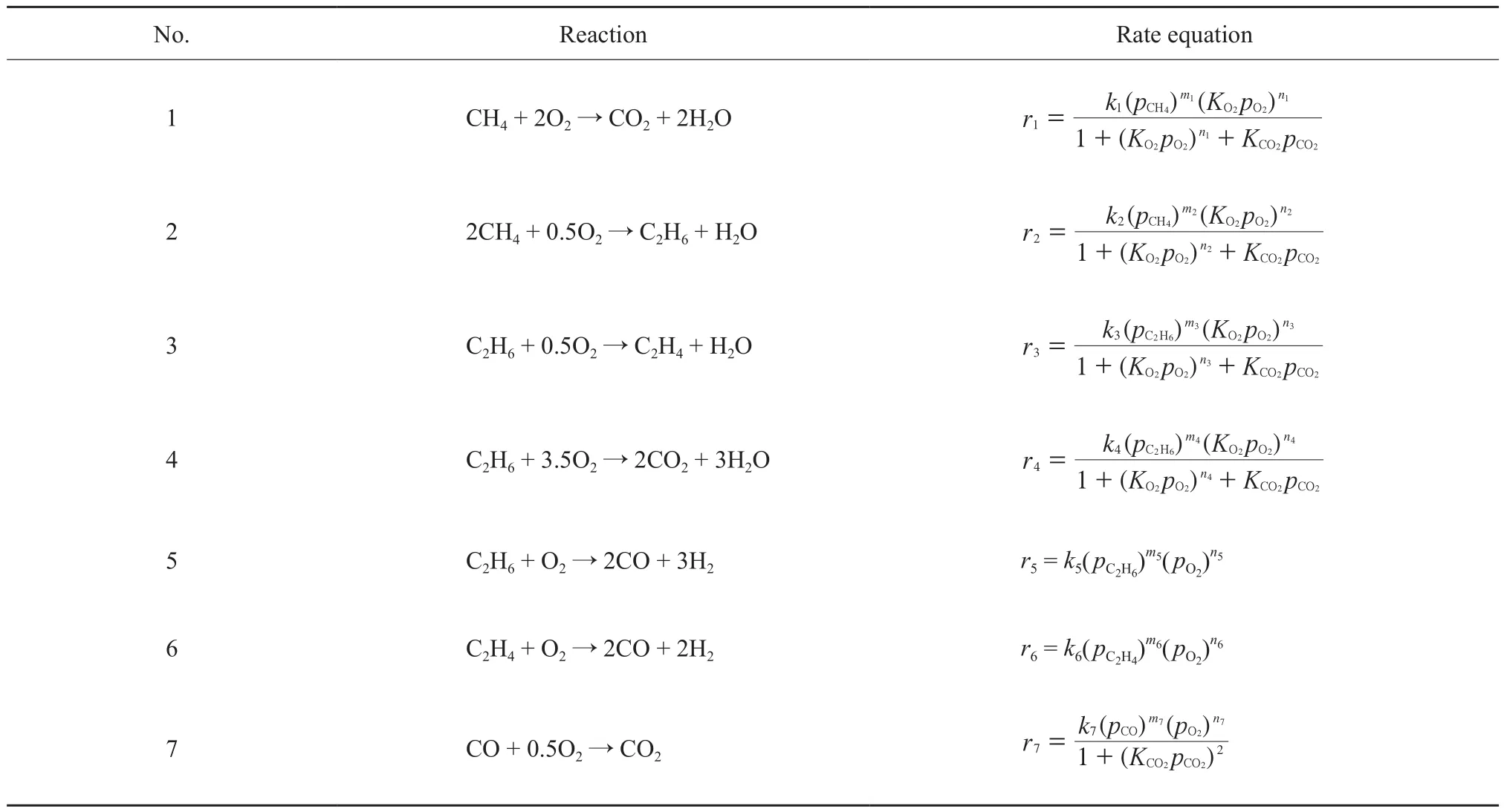
表 2 Sylvie Lacombe模型[27]Table 2 Sylvie Lacombe model[27]

表 3 Stansch模型[20]Table 3 Stansch model[20]

表 4 Kamali Shahri模型[32]Table 4 Kamali Shahri model[32]
2.5 Power-law模型
Tiemersma 等[18]以 Mn/Na2WO4/SiO2為催化劑,在O2分壓為0.7~28.2 kPa、甲烷進料分壓為 9.7~89.6 kPa、溫度800~900 ℃、進料流動速率在250 mL/min的條件下進行實驗,Power-law速率模型很好地描述了各反應步驟的速率方程(見表5),并將實驗結果與模擬結果進行對比。其他研究者也用Power-low速率模型很好地表示了各反應步驟,并對各步驟的參數進行了模擬[33]。
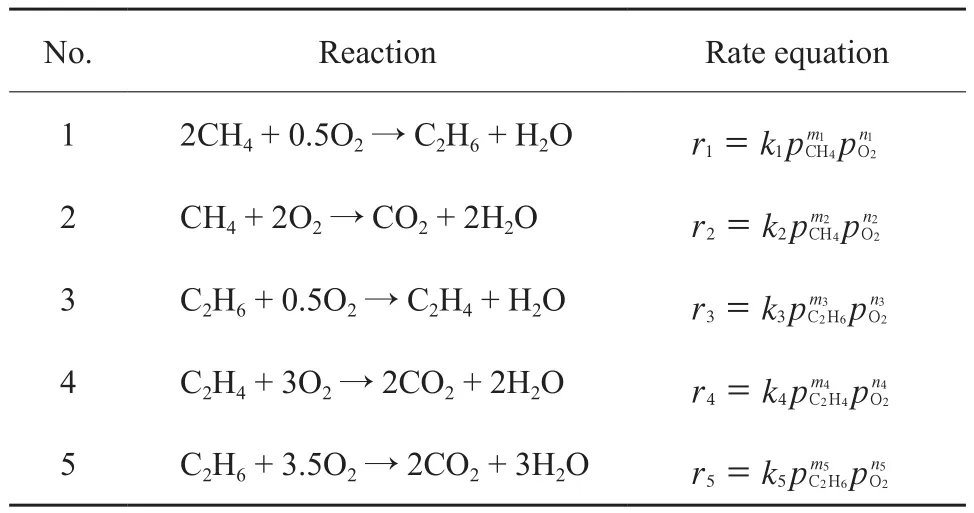
表 5 Power-law 模型[18]Table 5 Power-law model[18]
3 結語
甲烷氧化偶聯反應的反應機理較為復雜,文中介紹的4種機理模型均得到廣泛的應用,其中,Langmuir-Hinshelwood機理模型應用最為廣泛,但由于甲烷氧化偶聯反應的催化劑種類繁多,不同的催化劑在甲烷氧化偶聯反應中有不同的機理,需要研究者根據具體的催化劑具體分析。Stansch模型的反應動力學方程包含9個多相催化反應步驟和1個均相催化反應步驟,包含了甲烷氧化偶聯反應的大多數反應步驟,適用范圍廣。甲烷氧化偶聯制乙烯在科學研究中具有一定的科學意義和學術價值,是一門重要的研究課題。在催化劑反應研究中,催化劑動力學的研究一方面可以幫助了解催化劑的反應機理;另一方面也可為催化劑反應器的設計提供依據。鑒于目前許多催化劑的催化效果仍未達到工業生產的要求,研究者將催化劑的研究與催化劑動力學和反應機理的研究相結合,可進一步指導催化劑的研究。
符 號 說 明
Eaj活化能,kJ/mol
ΔHj吸附熱,kJ/mol
j -4,-3,-2,-1,0,1,2,3,4,5
K 吸附平衡常數,Pa-1
kj化學反應速率常數,mol/(g·s)
k0j指數前因子
mj反應級數
nj反應級數
p 壓力,Pa
R 氣體常數,J/(mol·K)
r 總包反應速率,mol/(g·s)
T 溫度,K
t 時間,s
θ 被吸附的活性位點
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
光學精密工程(2016年6期)2016-11-07 09:07:19
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
