四例頜面部木村病臨床及病理分析
2018-10-27 02:32:52郎小喬孫勇虎盧憲梅張福仁
中國麻風皮膚病雜志 2018年10期
郎小喬 孫勇虎 盧憲梅 張福仁
木村病(Kimura’s disease,KD)是一種以皮下軟組織無痛性包塊為主要臨床表現的慢性炎癥性疾病。本病較少見,臨床表現不典型,常與其他疾病相混淆,現將我院2017年1月1日至2018年5月31診療的4例木村病患者匯總報道如下。
1 臨床資料
患者1,女,14歲。1年前無明顯誘因在右眼瞼下出現腫塊,伴明顯瘙癢。體格檢查:右眼瞼下皮下腫塊,約2 cm×2 cm大,正常膚色,表面光滑,無觸痛(圖1a)。實驗室檢查:外周血嗜酸粒細胞略升高為0.53×109/L[(0.05~0.5)×109/L],嗜酸粒細胞比率升高為8.41%(0.5%~5%),IgE升高為151.1 ku/L(0~100 ku/L),其余指標未見明顯異常。右眼瞼下包塊組織病理學檢查(切至包塊):表皮大致正常,真皮中深層有較多淋巴細胞、嗜酸粒細胞浸潤,見淋巴濾泡(圖2a)。診斷:木村病。
患者2,女,66歲。20年前無明顯誘因出現左耳后腫物,七年前右腮部出現緩慢增大腫物,伴略微瘙癢。曾在當地診所不規律使用激素治療(劑量不詳),病情反復。體格檢查:右腮可見約8.5 cm×6.5 cm大的腫物,質地硬,活動度差,表面散在分布抓痕,無觸痛(圖1b)。實驗室檢查:血常規嗜酸粒細胞升高為5.43×109/L,嗜酸粒細胞比率升高為33.81%,IgE升高為1016.6 ku/L,其他指標未見明顯異常。右腮包塊組織病理學檢查(切至包塊):表皮大致正常,真皮增厚,纖維組織增生,散在較多嗜酸粒細胞及少量淋巴細胞、漿細胞浸潤,未見典型淋巴濾泡(圖2b)。診斷:木村病。
患者3,男,61歲。面部無明顯誘因出現多個結節2年,逐漸增大,偶有瘙癢。皮膚科檢查:雙側面頰散在分布花生粒大暗紅色皮下腫塊,無破潰,無觸痛(圖1c)。實驗室檢查:外周血嗜酸粒細胞為0.32×109/L,嗜酸粒細胞比率為2.8%,IgE升高為264.3 ku/L,其他指標未見明顯異常。面部包塊組織病理學檢查(切至包塊):表皮大致正常,真皮深淺血管周圍淋巴樣細胞和嗜酸粒細胞浸潤,真皮深部見多個濾泡樣結構,膠原纖維略增生(圖2c)。診斷:木村病。
患者4,男,45歲。面部腫塊1.5年,偶有瘙癢。體格檢查:面頰、耳后有膚色腫塊突出皮膚表面,有浸潤,左耳后腫塊觸診質硬(圖1d)。實驗室檢查:外周血嗜酸粒細胞為0.38×109/L,嗜酸粒細胞比率為5%,IgE升高為1200.4 ku/L,其他指標未見明顯異常。右頰包塊組織病理學檢查(切至包塊):表皮角化過度,真皮深層見淋巴濾泡樣結構形成,濾泡周圍淋巴細胞、嗜酸粒細胞浸潤(圖2d)。診斷:木村病。
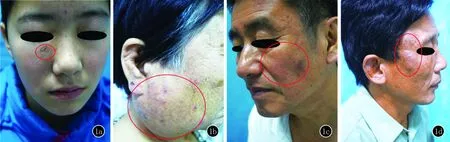
1a:右眼瞼下約2 cm×2 cm大皮下腫塊,正常膚色,表面光滑;1b:右腮可見巨大腫物,約8.5 cm×6.5 cm大,表面散在分布抓痕;1c:面頰散在分布暗紅色皮下腫塊,花生粒大,無破潰;1d:面頰有膚色腫塊突出皮膚表面,有浸潤
圖1 患者臨床圖片
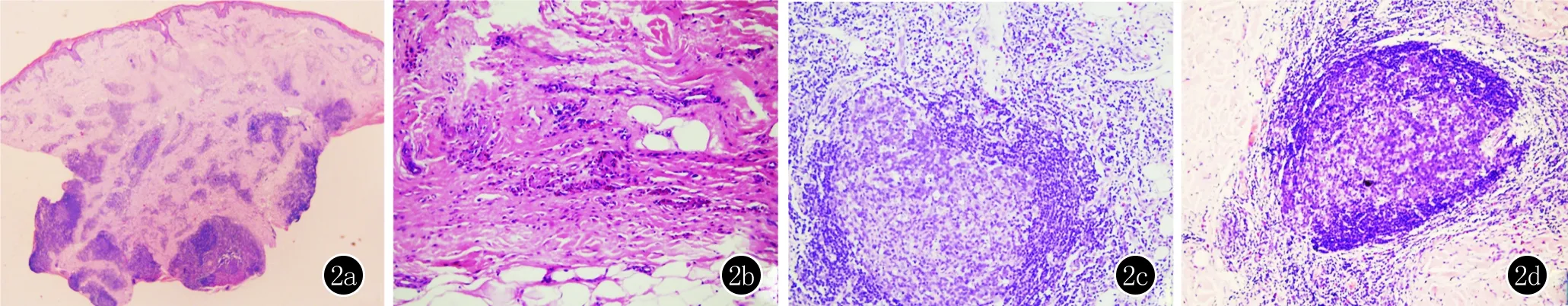
2a:真皮中深層有較多淋巴細胞、嗜酸粒細胞浸潤,見淋巴濾泡(HE,×20);2b:真皮深層散在較多嗜酸粒細胞及少量淋巴細胞、漿細胞浸潤,未見典型淋巴濾泡(HE,×200);2c:真皮深層可見典型濾泡樣結構(HE,×200);2d:可見清晰的淋巴濾泡結構,周圍有淋巴細胞、嗜酸粒細胞浸潤(HE,×200)
圖2 患者組織病理
2 討論
木村病由中國醫生金顯宅首次在1937年報道[1],后日本學者Kimura[2]在1948年進行詳細的闡述,故以其名字命名該病。這是一種少見的慢性良性炎癥性疾病,主要表現為頭頸部區域皮下軟組織無痛性腫塊,病因及發病機制尚不明確,可能是由于免疫調節損傷或持續性抗原刺激引起的特應性反應,如節肢動物叮咬、病毒感染、外傷、腫瘤和念珠菌感染等[3]。該病好發于亞裔30~50歲中青年男性, 2015年胡袒等[4]總結了國內82例患者信息,男女比例為3.3∶ 1,本文4例患者中男女比例為1∶1,年齡14~66歲,平均46.5歲,病程1~20年。性別比例與文獻不符的原因可能為樣本量過少。
臨床上,木村病多見于頭頸部區域,包塊質地較韌,大小不等,多數無明顯感覺,可有色素沉著和輕微瘙癢,少數伴有明顯瘙癢或壓痛。常累及耳周、腮腺、下頜下腺等區域,其次可見于腹股溝、四肢、腋窩等處,偶見于口腔、鼻咽、胸背部、縱隔等部位。約42%~100%的木村病患者會伴有淺表淋巴結腫大[5],常累及頦下、下頜下、頸部、腹股溝、腋窩淋巴結。本文4例患者有木村病常見的臨床表現,如4例患者腫物均發生在頭頸部區域,均為無痛性皮下腫物。同時也表明了該病少見的臨床表現,例如患者1發病年齡為13歲,該病發生于兒童患者中少見。有15%~18%的患者常合并腎臟疾病,其中有約2/3的患者為腎病綜合征[6],腎病癥狀可以在木村病診斷之前或之后出現[7],但二者之間的關系目前不明,有學者認為腎臟損害可能是由于免疫復合物介導的損傷,例如白介素、細胞因子、輔助T細胞免疫應答[8]。
實驗室檢查中,85.37%的患者外周血嗜酸性粒細胞計數升高,血清IgE水平大多也會升高,約占42.68%[4]。本文患者中有2例外周血嗜酸粒細胞相關指標和血清IgE水平均升高,有2例僅血清IgE水平升高,而外周血嗜酸性粒細胞相關指標正常。與文獻中數據差距大的可能原因是本文樣本量少。組織病理學表現主要為形成大小不等的淋巴濾泡,生發中心增生活躍,濾泡間區有嗜酸粒細胞浸潤,可形成嗜酸性微膿腫,毛細血管后微靜脈增生。本文4例患者均有濾泡間嗜酸粒細胞浸潤,3例可見典型的淋巴濾泡。
木村病常與嗜酸粒細胞增多性血管淋巴樣增生(ALHE)常混淆,ALHE由Wells和Whimster在1969年首次報道[9],二者都多見于頭頸部區域,ALHE是一種罕見的良性腫瘤,多見于女性,臨床上多表現為頭頸部區域皮膚2~3 cm大的淺棕色丘疹或結節。通常情況下血清IgE在正常范圍內,外周血嗜酸粒細胞計數小于白細胞總數的10%。其組織病理變化雖與木村病的具有一定相似性,但ALHE 病變常只侵犯淺層皮膚,病變范圍小,一般不累及淋巴結或腮腺,而木村病浸潤深度更深在、受累范圍更為廣泛。此外,霍奇金淋巴瘤和髓細胞性白血病也會出現嗜酸性粒細胞升高,分別通過淋巴結活檢和骨髓活檢可以與木村病相鑒別。木村病可發生在軀體任何部位,有學者報道一例發生在陰囊處的木村病,要與陰囊腫瘤、陰囊結核和腹股溝疝鑒別[10]。該病臨床表現不典型,可以肺栓塞為首發癥狀,嗜酸粒細胞增多可能是血栓形成的原因,但要排除其它導致血栓的可能因素[5]。
木村病目前尚無統一的治療方案,系統性運用糖皮質激素可以有效控制病情,但容易復發。有學者認為手術完全切除是一線治療方法,但復發率仍高達25%[5]。另有免疫抑制劑環孢菌素可以有效控制病情,放射治療可以用于治療復發或病灶持久的患者。當合并腎臟并發癥時治療更為棘手,有學者建議此類病人在進行內科治療之前可以優先進行手術切除治療[11]。本文4例患者中有2例在本院接受治療,其余2例患者確診后回轉外院治療。患者1給予口服醋酸潑尼松片,鹽酸雷尼替丁膠囊,碳酸鈣D3片(鈣爾奇D),局部外用0.03%他克莫司軟膏(明之欣)。14周后外周血嗜酸粒細胞0.17×109/L,嗜酸粒細胞比率為2.4%,IgE為81.7 ku/L,均降至正常。隨訪16個月,未見復發。患者2首次治療予口服甲潑尼龍片32 mg/d、靜脈滴注甲氨蝶呤15 mg/w。第二月復查見右腮腫物消失,甲潑尼龍減量至24 mg/d,甲氨蝶呤改為口服15 mg/w。后因患者產生了激素不良反應(眼睛不適),因此停用糖皮質激素,維持復方甘草酸苷片、甲氨喋呤片口服15 mg/w治療,隨訪9個月,未見復發。
總之,木村病沒有統一的診斷標準,典型癥狀為:頭頸部無痛性皮下包塊,嗜酸性粒細胞顯著增多,IgE升高。該病為良性病變,但易復發,一般無惡變傾向,主要依靠病理學確診。由于全球范圍內少見,其發病機制不清,目前治療手段尚不成熟,統一的治療方案需進一步摸索。
