超高效液相色譜-四極桿飛行時間質譜法快速篩查水產品中16種激素殘留
2018-10-31 00:52:28陳秋華張天聞黃東仁
食品科學 2018年20期
關鍵詞:方法
陳秋華,張天聞*,傅 紅,3,黃東仁
(1.福州大學生物科學與工程學院,福建 福州 350108;2.福建省海洋環境與漁業資源監測中心,福建 福州 350003;3.福州大學 福建省海洋酶工程重點實驗室,福建 福州 350108)
激素是指由體內的某一細胞、腺體或者器官所產生的可以影響機體內其他細胞活動的化學物質,主要包括性激素、孕激素和糖皮質激素等[1]。激素具有提高飼料轉化率、縮短動物生長周期、促進動物生長繁殖等作用,因此存在在水產養殖中違規使用以提高經濟效益的現象[2-5]。研究表明,食品中激素的殘留會導致性早熟、乳腺癌及前列腺癌發病率提高[6-7]。2002年我國農業部第235號公告中已禁止使用諾龍、甲基睪丸酮、群勃龍等激素物質,并規定這些化合物在動物性食品中不得檢出[8]。同時歐盟第96/22/EC指令、美國食品藥物管理局也禁止在動物源性食品中使用激素類藥物[9]。
目前激素類藥物的殘留檢測方法主要有高效液相色譜法[10]、液相色譜-串聯三重四極桿質譜法[11-14]、超高效液相色譜-四極桿飛行時間質譜法[15-18]等。其中液相色譜法無法區分保留時間相同或相近的物質,因而定性能力較差。液相色譜-串聯三重四極桿質譜法憑借多反應監測模式,其靈敏度和定性能力都較高,是目前主要的檢測方法,但受分辨率、掃描速率及分析模式的限制,無法實現高通量快速篩查分析[19]。液相色譜-四極桿飛行時間質譜法具有高分辨率、高通量、靈敏度高的特點,可實現復雜食品基質中目標物的定性分析和高通量快速篩查分析,同時還可通過建立篩查譜庫實現在無標準品情況下對目標物進行篩查和確認[20-23]。
QuEChERS(Quick,Easy,Cheap,Effective,Rugged and Safe)是由Anastassiades等[24]于2003年建立的樣品前處理技術,具有操作簡單、分析速度快、分析范圍廣等優點,此方法已廣泛應用于食品中農藥殘留的檢測,近年來也逐漸被用于動物性食品中獸藥殘留的檢測[25-27]。熊雯等[28]建立了檢測水產品中18 種磺胺類藥物的QuEChERS-液相色譜-四極桿飛行時間質譜方法。朱萬燕等[29]建立了快速檢測豬肉中33 種獸藥殘留的QuEChERS-液相色譜-四極桿飛行時間質譜方法。但目前采用QuEChERS-液相色譜-四極桿飛行時間質譜方法同時對多種水產品基質中的激素殘留進行篩查分析的文獻報道還較少。本實驗通過超高效液相色譜-四極桿飛行時間質譜(ultra performance liquid chromatographyquadrupole time of fl ight mass spectrometry,UPLC-Q-TOF MS)技術建立篩查數據庫,同時采用改進的QuEChERS方法結合UPLC-Q-TOF MS技術建立水產品中激素快速、有效的篩查方法,以期為水產品中激素殘留風險監測提供有效技術手段,同時為水產品中獸藥殘留的篩查分析提供借鑒。
1 材料與方法
1.1 材料與試劑
勃地酮、甲睪酮、睪酮、諾龍、美雄酮、表睪酮、群勃龍、17α-羥基孕酮、甲羥孕酮乙酸酯、乙酸氯地孕酮、21α-羥基孕酮、乙酸甲地孕酮、醋酸美倫孕酮、孕酮、氫化可的松、潑尼松、睪酮-D3(純度均大于93.6%) 德國Dr. Ehrenstorfer公司;甲醇、乙腈、乙酸乙酯、正己烷、丙酮(均為色譜純) 德國Merck公司;甲酸(色譜純) 美國Scientific公司;乙酸(色譜純)美國Tedia公司;無水硫酸鎂(分析純) 國藥集團化學試劑有限公司;氯化鈉(農殘級) 上海安譜公司。
1.2 儀器與設備
ACQUITY H-CLASS型UPLC儀、Xevo G2-S型Q-TOF MS儀 美國Waters公司;V-700旋轉蒸發儀德國Heidolph公司;3-30K高速冷凍離心機 德國Sigma公司;MS3 digital旋渦振蕩器 德國IKA公司;multi Reax振蕩器 德國Heidolph公司;Milli-Q超純水系統美國Millipore公司。
1.3 方法
1.3.1 標準溶液的配制
分別準確稱取適量標準物質或同位素內標物質,用甲醇溶解并定容至100 mL,配制成100 μg/mL的單標儲備液,避光保存于-18 ℃冰箱。準確移取適量標準儲備液用甲醇定容為500 ng/mL的混合標準儲備液,保存于4 ℃冰箱。用甲醇-水體積比35∶65稀釋混合標準儲備液獲得1、5、10、25、50、100 μg/L的標準工作液,其中標準工作液中均含有20 μg/L的同位素內標睪酮-D3,將標準工作液保存于4 ℃冰箱。
1.3.2 樣品前處理
準確稱取2.00 g樣品(精確至0.01 g)于50 mL離心管中,加入80 μL 500 ng/mL的激素混標溶液,靜置15~20 min,加入10 mL 1%乙酸-乙腈溶液,渦旋混勻1 min,加入鹽析劑(2 g無水硫酸鎂和1 g氯化鈉),渦旋混勻1 min,在4 ℃、10 000 r/min離心5 min,準確吸取上清液5 mL于新的15 mL離心管中,加入3 mL正己烷,渦旋混勻1 min,在4 ℃、5 000 r/min離心5 min,棄去正己烷層。再往離心管中加入凈化劑(800 mg無水硫酸鎂和150 mg C18(南美白對蝦、大黃魚、草魚基質)或200 mg C18(梭子蟹基質)),渦旋混勻2 min,在4 ℃、5 000 r/min離心10 min,準確移取3 mL上清液于25 mL雞心瓶中。38 ℃旋蒸至干,用1 mL含20 ng/mL睪酮-D3的定容液(甲醇-水體積比35∶65)復溶,過0.22 μm有機濾膜,待上機測定。
1.3.3 UPLC條件
色譜柱:Waters Acquity UPLC BEH C18柱(3.0 mm×100 mm,1.7 μm);柱溫40 ℃;樣品室溫度10 ℃;進樣體積10 μL;流動相:A為0.1%甲酸溶液,B為甲醇;流速0.3 mL/min;梯度洗脫條件:0~1 min,35% B;1~5 min,35%~65% B;5~8 min,65%~80% B;8~11 min,80%~98% B;11~14 min,98% B;14~15 min,98%~35% B;15~18 min,35% B。
1.3.4 MS條件
離子源:電噴霧離子源;電離模式:正離子;檢測模式:MSE;質量數采集范圍:50~1 000 Da;毛細管電壓:3 kV;離子源溫度:120 ℃;脫溶劑氣溫度:420 ℃;脫溶劑氣流速:800 L/h;錐孔氣流速:50 L/h;錐孔電壓:40 V;LockSpray程序:采用亮氨酸腦啡肽(1 μg/mL,m/z 556.277 1)每10 s切換一次對質量數進行實時校正。
1.3.5 數據庫的建立
本實驗在MSE全掃描模式下,在低能量碰撞通道獲得目標物的保留時間、母離子精確質量數等信息,在高能量碰撞通道獲得目標物的子離子精確質量數。在MS/MS模式下,通過相應碰撞能量下的離子碎片信息進一步確證。借助Waters公司Masslynx 4.1軟件中的fragment軟件分析化合物的結構裂解信息。最后通過Chromalynx xs數據庫軟件,建立包含16 種激素名稱、分子式、保留時間、精確質量數、子離子等信息的篩查數據庫。具體的數據庫信息如表1所示,色譜圖見圖1。
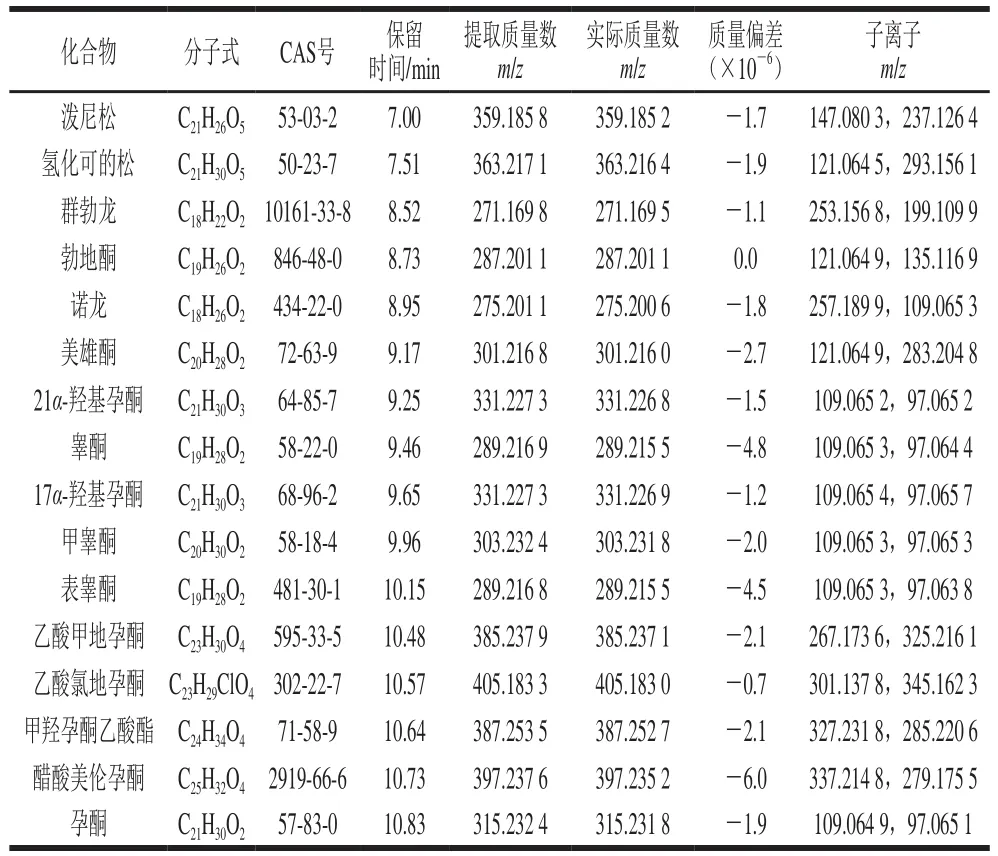
表1 激素的分子式、CAS號、保留時間、提取質量數、子離子Table 1 Molecular formula, CAS No., retention time, m/z and product ion of 16 hormones

圖1 16 種激素的色譜提取圖Fig. 1 Extracted ion chromatogram of 16 hormones
1.3.6 基質效應的計算
基質效應的消除方法主要有內標法、基質匹配等。基質效應是通過比較目標化合物在純溶劑與在基質中的峰面積進行評估的,其計算公式如下:

1.4 數據處理
在Chromalynx xs軟件中,運用建立好的數據庫對UPLC-Q-TOF MS采集的樣品數據進行檢索匹配分析,利用質譜軟件的自動去卷積功能,并與數據庫的保留時間、精確母離子質量數、子離子質量數等相關參數進行對比,當分析物與譜庫中化合物的保留時間偏差小于±0.2 min,母離子和子離子質量數偏差小于±3 mu時,顯示為陽性結果,隨后針對陽性化合物使用targetlynx軟件進行定量分析。與MS/MS相比,Q-TOF具有更快的掃描速度和更高的質量精度,從而可實現高通量篩查,另外本方法構建的是一個開放的體系,待測樣品一次進樣,經Q-TOF全掃描獲得的全譜數據可多次使用,后期可通過不斷增加數據庫中的化合物來擴展對原樣品數據的篩查范圍。
2 結果與分析
2.1 MS條件優化結果
為獲得最佳的電離效率、提高靈敏度,本實驗以100 μg/L的混合標準溶液對錐孔電壓、離子源溫度、脫溶劑氣溫度、脫溶劑氣流速等質譜參數進行優化,根據總離子流響應值和各化合物的分子離子峰響應值的強度確定最佳質譜參數。以錐孔電壓為例,錐孔電壓太小會降低檢測靈敏度,太大則會造成離子源內裂解,本實驗研究了16 種激素在錐孔電壓為30~100 V時分子離子峰的響應情況,實驗發現當錐孔電壓為42 V時,激素總體響應值最高,同理對質譜的離子源溫度(100~150 ℃),去溶劑化溫度(350~500 ℃)和脫溶劑氣流速(500~1 000 L/h)進行優化,優化后的質譜參數見1.3.4節。
2.2 QuEChERS樣品前處理的優化
2.2.1 提取方法的優化結果
本實驗考察乙腈、1%乙酸-乙腈溶液、乙酸乙酯和丙酮4 種提取劑的效果。如圖2所示,分別用南美白對蝦、大黃魚和梭子蟹為基質,1%乙酸-乙腈溶液作為提取劑時16 種激素的平均回收率分別為83.3%、81.4%和83.8%,均高于乙腈、乙酸乙酯和丙酮,因而本實驗選用1%乙酸-乙腈溶液作為提取溶劑。實驗中還發現,乙酸乙酯和丙酮作為提取劑時,共提物較多,增加了提取液后期的凈化難度,有乙腈存在的體系中共提物較少,這可能因為乙腈具有沉淀蛋白質的作用,有利于后期的凈化。
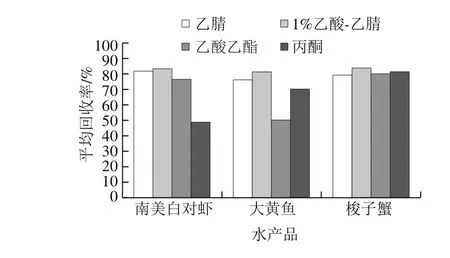
圖2 不同提取劑對16 種激素回收率的影響Fig. 2 Effects of different extraction solvents on the recoveries of 16 hormones
2.2.2 凈化方法的優化結果
QuEChERS方法常用的吸附劑有乙二胺-N-丙基硅烷(primary secondary amine,PSA)、十八烷基鍵合硅膠吸附劑(C18)、中性氧化鋁(Alu-N)、石墨化碳黑(graphitized carbon blacks,GCB)等,其中C18和Alu-N主要用于去除樣品中的脂肪、脂類等非極性有機物,而PSA和GCB主要用于去除樣品中的色素、有機酸和糖類等物質。本實驗分別考察以PSA、C18、Alu-N以及PSA-Alu-N、PSA-C18、PSA-C18-GCB作為凈化劑時的凈化效果,南美白對蝦、大黃魚、梭子蟹基質的凈化效果如圖3~5所示。實驗結果表明,以C18吸附劑作為凈化劑時16 種激素凈化效果最好,在南美白對蝦、大黃魚和梭子蟹基質中,其回收率分別在61.9%~93.9%、64.2%~119.7%、55.6%~112.5%范圍內。因此本實驗采用選擇C18作為吸附劑。本實驗還進一步對C18的用量進行優化,對比了100、150、200、250、300 mg C18吸附劑對目標化合物回收率的影響,實驗結果如圖6所示,在南美白對蝦、大黃魚和梭子蟹基質中,C18吸附劑加入量分別為150、150、200 mg時凈化效果最好,16 種激素平均回收率分別為83.5%、85.6%、84.0%。其中C18吸附劑加入量過低或過高都會使回收率降低,這可能是由于C18吸附劑加入量不夠時,無法凈化充分;而加入量過多時,吸附劑本身對激素也存在一定的吸附作用。
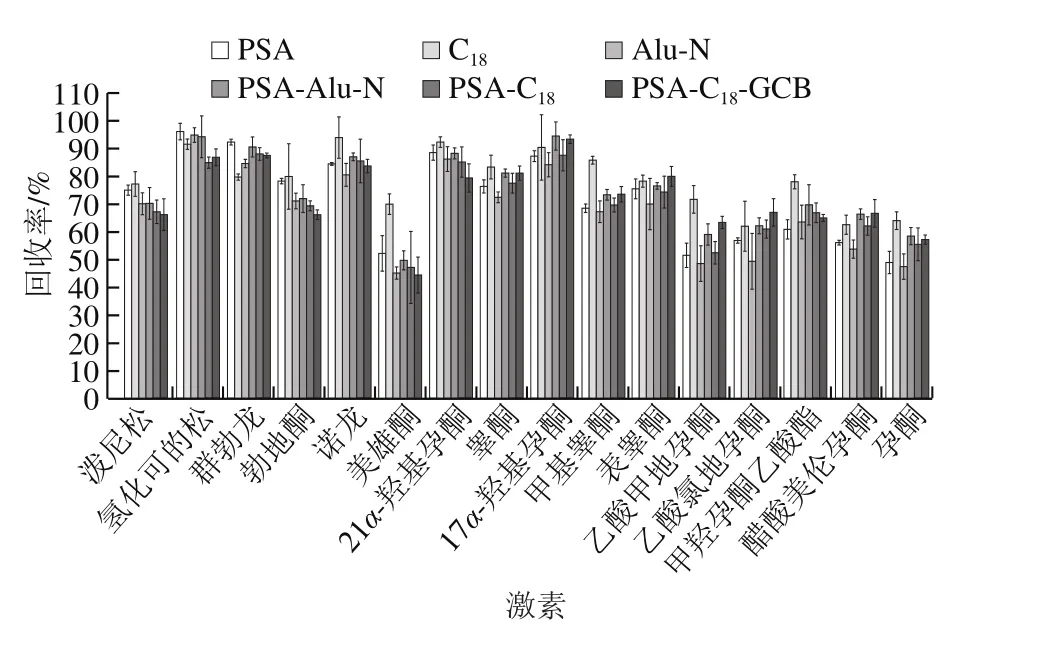
圖3 南美白對蝦基質中不同吸附劑對16 種激素的回收率的影響Fig. 3 Effects of different sorbents on the recoveries of 16 hormones in Penaeus vannamei matrix
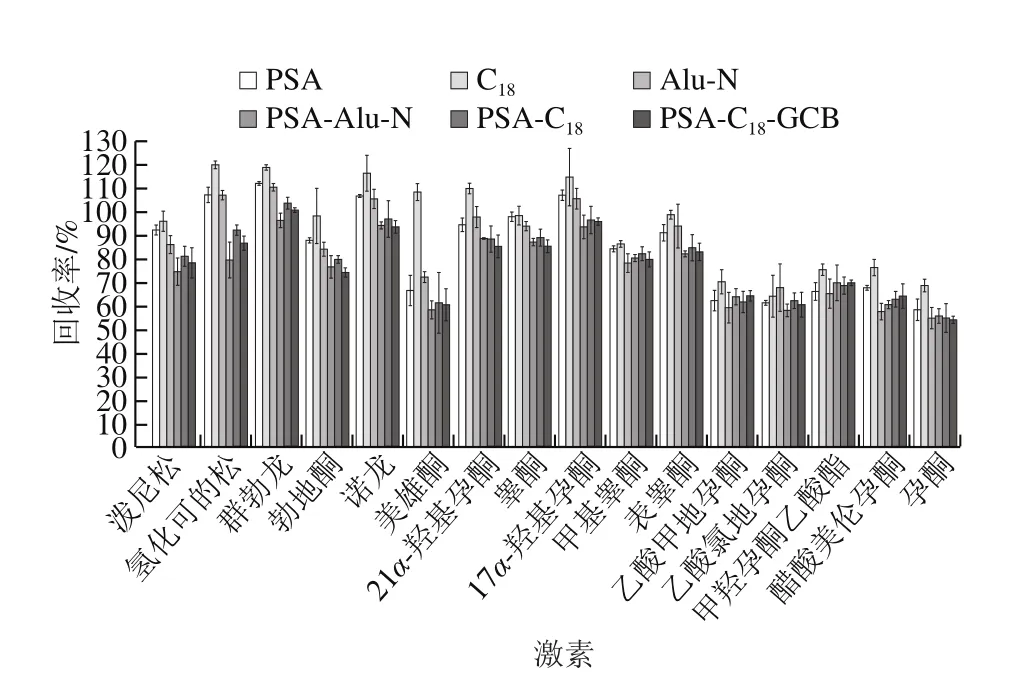
圖4 大黃魚基質中不同吸附劑對16 種激素的回收率的影響Fig. 4 Effects of different sorbents on the recoveries of 16 hormones in Pseudosciaena crocea matrix

圖5 梭子蟹基質中6 種吸附劑對16 種激素的回收率的影響Fig. 5 Effects of different sorbents on the recoveries of 16 hormones in swimming crab matrix

圖6 C18吸附劑添加量對16 種激素回收率的影響Fig. 6 Effects of C18 amount on the recoveries of 16 hormones
2.3 方法學驗證結果
2.3.1 基質效應
當基質效應在-20%~20%范圍內時,基質效應不明顯,超過20%時,表現為明顯的基質增強效應,低于-20%時,表現為明顯的基質抑制效應[30]。如表2所示,在南美白對蝦基質中,16 種激素的基質效應均在±20%范圍內,基質效應不明顯;在大黃魚基質中,勃地酮、美雄酮、潑尼松表現為明顯的基質抑制效應;在梭子蟹基質中,美雄酮表現為明顯的基質抑制效應,氫化可的松表現為明顯的基質增強效應;在草魚基質中,勃地酮、美雄酮、表睪酮、潑尼松表現為明顯的基質抑制效應。為獲得更準確的結果,本實驗采用通過基質匹配溶液的方法對基質效應進行校正。
2.3.2 線性范圍、檢出限和定量限
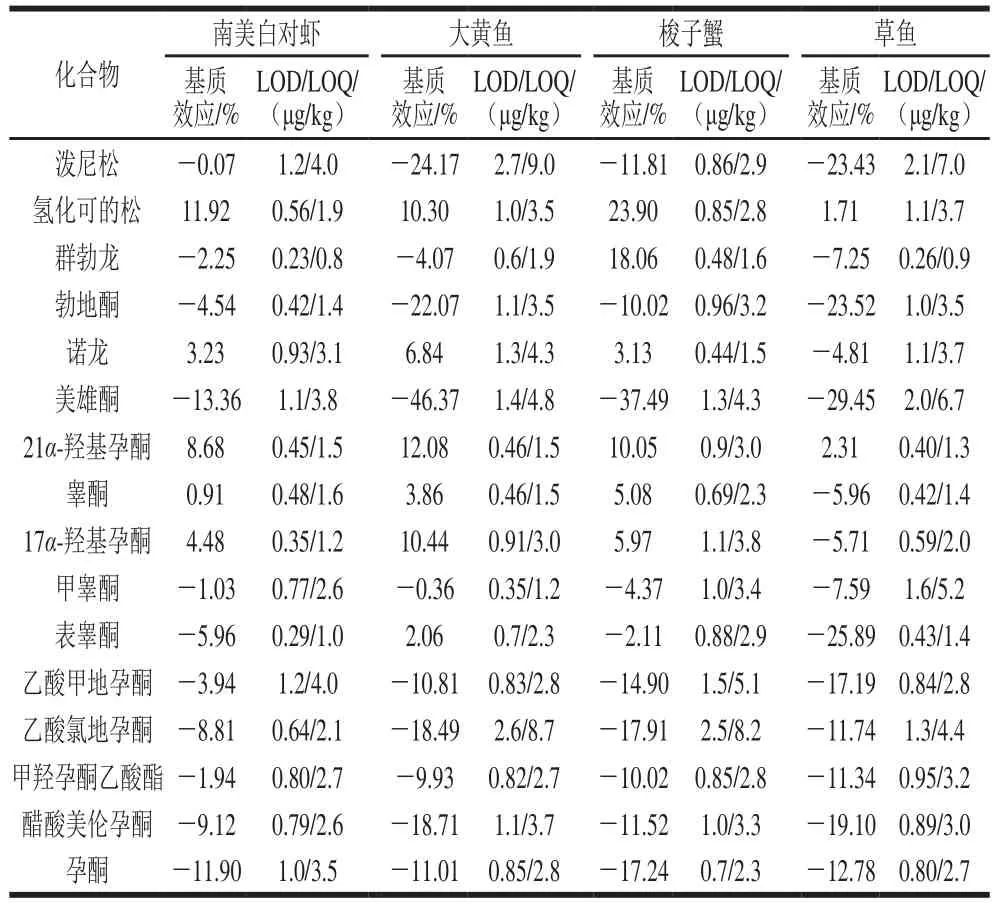
表2 激素的基質效應、方法LOD和LOQTable 2 Matrix effects, LODs and LOQs for all matrices tested
將4 種空白基質樣品按1.3.2節處理后,添加不同質量濃度(1、5、10、25、50、100 μg/L)的混合標準溶液,以各激素母離子的峰面積與同位素內標的峰面積的比值(Y)為縱坐標,基質標準溶液的質量濃度(X)為橫坐標,繪制標準曲線。各激素在1~100 μg/L范圍內線性關系良好,16 種激素在南美白對蝦、大黃魚、梭子蟹、草魚4 種基質中的基質匹配標準曲線的相關系數(r2)分別為0.994 5~0.999 9、0.993 8~0.999 9、0.990 6~0.999 9、0.996 4~0.999 6。
以3 倍信噪比為檢出限(limit of detection,LOD),10 倍信噪比為定量限(limit of quantitation,LOQ)。如表2所示,16 種激素在南美白對蝦基質中的方法LOD為0.2~1.2 μg/kg,LOQ為0.8~4.0 μg/kg;在大黃魚基質中的方法LOD為0.4~2.7 μg/kg,LOQ為0.8~4.0 μg/kg;在梭子蟹基質中的方法LOD為0.4~2.5 μg/kg,LOQ為1.5~8.2 μg/kg;在草魚基質中的方法L O D為0.3~2.1 μ g/k g,LOQ為0.9~7.0 μg/kg。
2.3.3 回收率和精密度
在10、20、50 μg/kg三個添加水平下做回收率實驗,以考察方法的準確性和精密度(表3),16 種激素在南美白對蝦基質中的回收率為64.1%~102.8%,相對標準偏差(relative standard deviation,RSD)為0.9 3%~7.6%;在大黃魚基質中的回收率為65.2%~98.6%,RSD為1.9%~10.9%;在草魚基質中的回收率為68.8%~114.4%,RSD為1.2%~9.2%;在梭子蟹基質中,除孕酮和潑尼松外,其余14 種激素的回收率為60.5%~121.0%,RSD為1.6%~10.4%,孕酮和潑尼松的回收率分別為53.6%~60.8%和91.7%~135.2%,RSD分別為2.3%~4.5%和2.7%~7.6%,這可能是由于梭子蟹基質中存在一些無法去除的雜質造成的基質抑制或基質增強效應,但本方法仍可保證二者的回收率處于相對穩定的狀態。總體上,本研究建立的方法可滿足水產品中16 種激素的實際檢測要求。
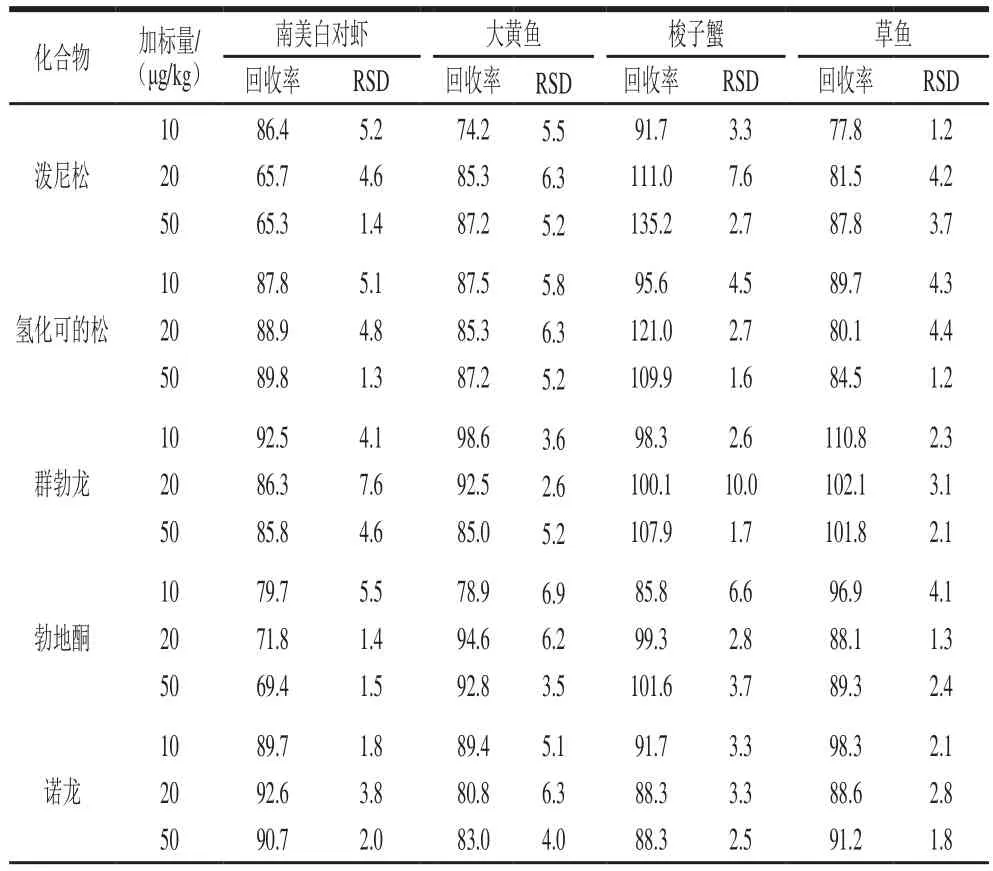
表3 激素類化合物的回收率和相對標準偏差(n=6)Table 3 Recovery and repeatability (RSD) for all matrices tested (n=6)%
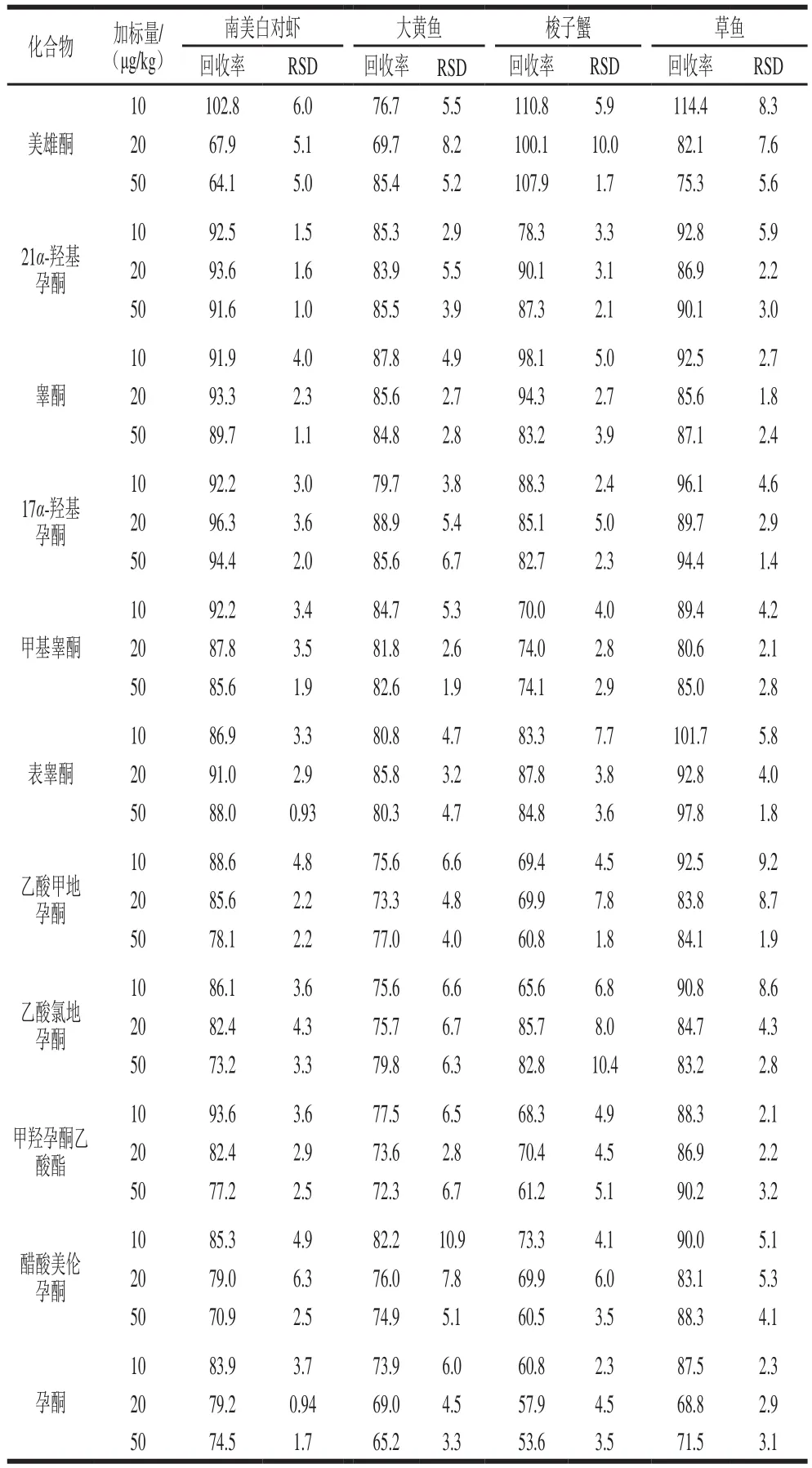
續表3
2.4 實際樣品檢測結果
采用本方法對36 份水產品樣品(南美白對蝦、大黃魚、草魚、鯉魚)進行檢測,其中大黃魚和南美白對蝦樣品均無檢出,6 份草魚樣品檢出氫化可的松,含量為5.9~204.1 μg/kg,8 份鯉魚樣品中檢出氫化可的松,含量為12.3~105.6 μg/kg。氫化可的松是天然存在的一種腎上腺糖皮質激素,同時也可由人工合成使用。孫利東等[31]對雞肉和牛奶中氫化可的松的本底值進行測定,其結果在0.06~0.43 μg/kg范圍內,這表明生物體內氫化可的松的本底值較低。但本次實際檢測樣品中檢出的氫化可的松含量相對較高,外用導致的可能性較大。這與李晴[32]、蔡惠堅[33]等分別在寶石斑魚、草魚中檢出氫化可的松的報道一致,表明在此類水產品中存在使用該激素的可能。目前我國農業部235號公告[8]規定允許在動物源食品中使用氫化可的松,且無最高殘留限量要求,而文獻[34]規定了牛奶中氫化可的松的最大殘留限量為10 μg/kg,表明氫化可的松仍有危害人體健康的可能,因此針對水產品中氫化可的松的殘留,也應提前開展相關的風險監測工作。
3 結 論
本實驗優化了QuEChERS前處理方法,并結合UPLCQ-TOF MS技術,建立快速篩查魚、蝦、蟹等水產品中16 種激素殘留的方法。本方法前處理簡單高效,目標激素回收率滿足篩查需求,同時結合高分辨質譜,使篩查結果更加準確可靠,是開展水產品中多激素殘留快速篩查的一種有效手段。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
