1例少見漿母細胞骨髓瘤的實驗室診斷及文獻復習
2018-11-05 09:27:16高曉娟王亞奇王寧寧岳保紅
檢驗醫學 2018年10期
李 佳, 高曉娟, 王亞奇, 王寧寧, 岳保紅
(鄭州大學第一附屬醫院檢驗科,河南 鄭州 450052)
漿細胞腫瘤是一類漿細胞系統異常增生所致的血液系統惡性腫瘤,包含多種亞型,其中漿母細胞骨髓瘤(plasmablastic plasma cell myeloma,PPCM)較為少見,僅見少數病例報道[1-2]。漿細胞瘤出現漿母細胞的分化常常說明其惡性程度較高,因此與其他具有漿母細胞形態的B細胞腫瘤的鑒別至關重要。臨床上常見的相似疾病有漿母細胞淋巴瘤(plasmablastic lymphoma,PBL)、間變性漿細胞骨髓瘤(anaplastic plasma cell myeloma,APCM)。本研究根據1例PPCM患者骨髓標本中所見的細胞形態學特征,并結合流式細胞術檢測免疫表型、臨床病史等作相關介紹,對PPCM的相關文獻進行復習。
1 病例資料
1.1 基本情況
患者,女,35歲,因“發現右側頸部包塊”入院。患者2月前無意間發現右側頸部包塊,無壓痛,無聲音嘶啞,無吞咽困難、飲食嗆咳等不適。既往無高血壓病、冠心病、糖尿病等病史,無結核、肝炎等傳染病史,無外傷史,無輸血、獻血史。查體可觸及右側頸部淋巴結,質硬,活動欠佳,未觸及甲狀腺。
1.2 血清學檢查
血常規(全血細胞分類):白細胞計數7.1×109/L,紅細胞計數3.53×1012/L,血小板計數170×109/L,血紅蛋白107 g/L。凝血功能:凝血酶原時間11.70 s,國際標準化比率1.04,纖維蛋白原5.61 g/L,D-二聚體1.935 μg/mL。乙型肝炎病毒、丙型肝炎病毒、梅毒、人類免疫缺陷病毒血清學檢測均為陰性。肝腎功能、電解質等檢查均未見明顯異常。血清免疫固定電泳:IgG 28.61 g/L,IgA 1.24 g/L,IgM 0.92 g/L,M蛋白陽性,IgE 13.26 IU/mL,κ/λ比值為12.47。
1.3 骨髓穿刺檢查
骨髓象:有核細胞增生明顯活躍,粒系占有核細胞的4.4%,紅系占0.4%,淋巴細胞占3.6%,主要成分為形態不能分類的異常細胞,占有核細胞的91.6%。該類細胞胞體偏大,呈現圓形、類圓形、不規則形,胞漿量中等,染藍色,胞漿邊緣不整齊,部分可見偽足,易見空泡。胞核圓形、類圓形,偶見雙核,核染色質細致,核仁隱顯不一[圖1(a)]。細胞髓過氧化物酶染色為陰性[圖1(b)],糖原染色呈粗顆粒狀陽性[圖1(c)]。瀏覽全片,在片尾易見此類成團細胞,同時易見吞噬細胞吞噬血小板、粒細胞、色素等噬血現象。
骨髓流式細胞術檢測結果:骨髓中發現占有核細胞20.67%的異常細胞,該類細胞表達CD38、CD138、CD71、Ki-67、CD45,不表達CD19、CD56、CD20、CD3、CD4、CD8、CD33、CD117等表面抗原(圖2),胞漿內輕鏈未見表達。FSC值較高提示細胞體積較大,SSC值較高可能與形態學上的細胞核形變異及胞漿內大量空泡有關。結合免疫表型及流式細胞術圖形,符合漿母細胞的表型特征。
1.4 影像及病理學檢查
超聲示:右側頸部多發低回聲包塊,考慮腫大淋巴結;肝、膽、胰、脾及雙腎未見明顯異常。正電子發射電子計算機斷層掃描(positron emission tomography-computed tomography,PET-CT):右側頭頸部、腹膜后、雙側髂血管旁多發腫大淋巴結代謝活躍,全身骨骼代謝活躍,考慮為腫瘤轉移。右頸部淋巴結活檢:CD3-、CD20-、CD21-、CD30-、EMA+、ALK-、Bcl-6-、MUM-1+、CD38+、CD138+、Bcl-2-、Ki-67 90%+、AE1/AE3-、CK5/6-、CK7-、CK8/18-、CK20-、CD56-、CD43-、EBER-,結合臨床病史及所有檢查確診為分化差的PPCM。
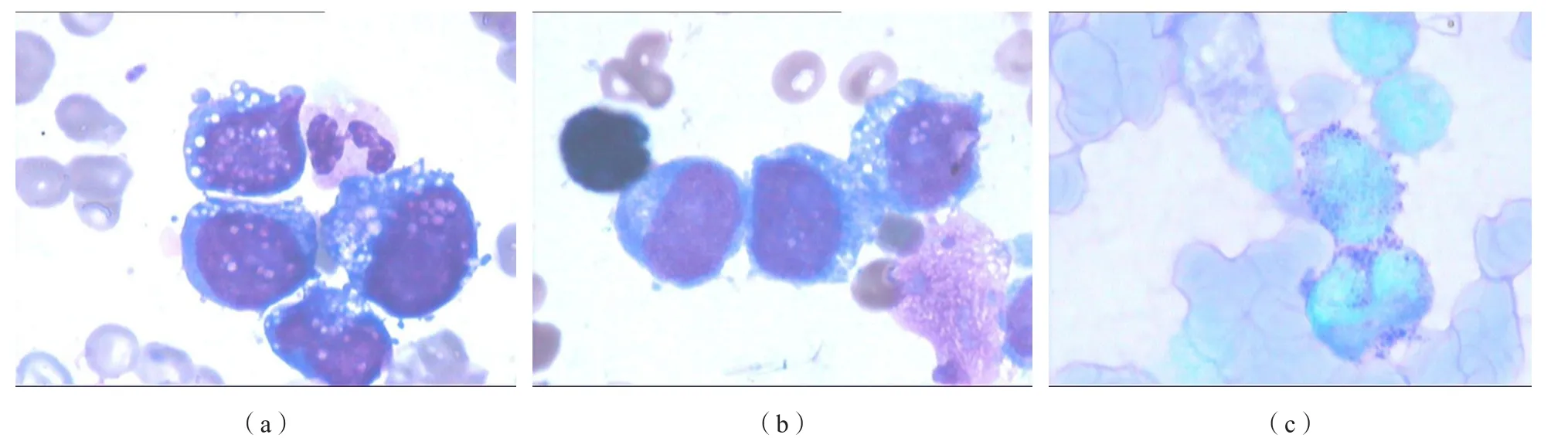
圖1 患者骨髓細胞形態化學染色
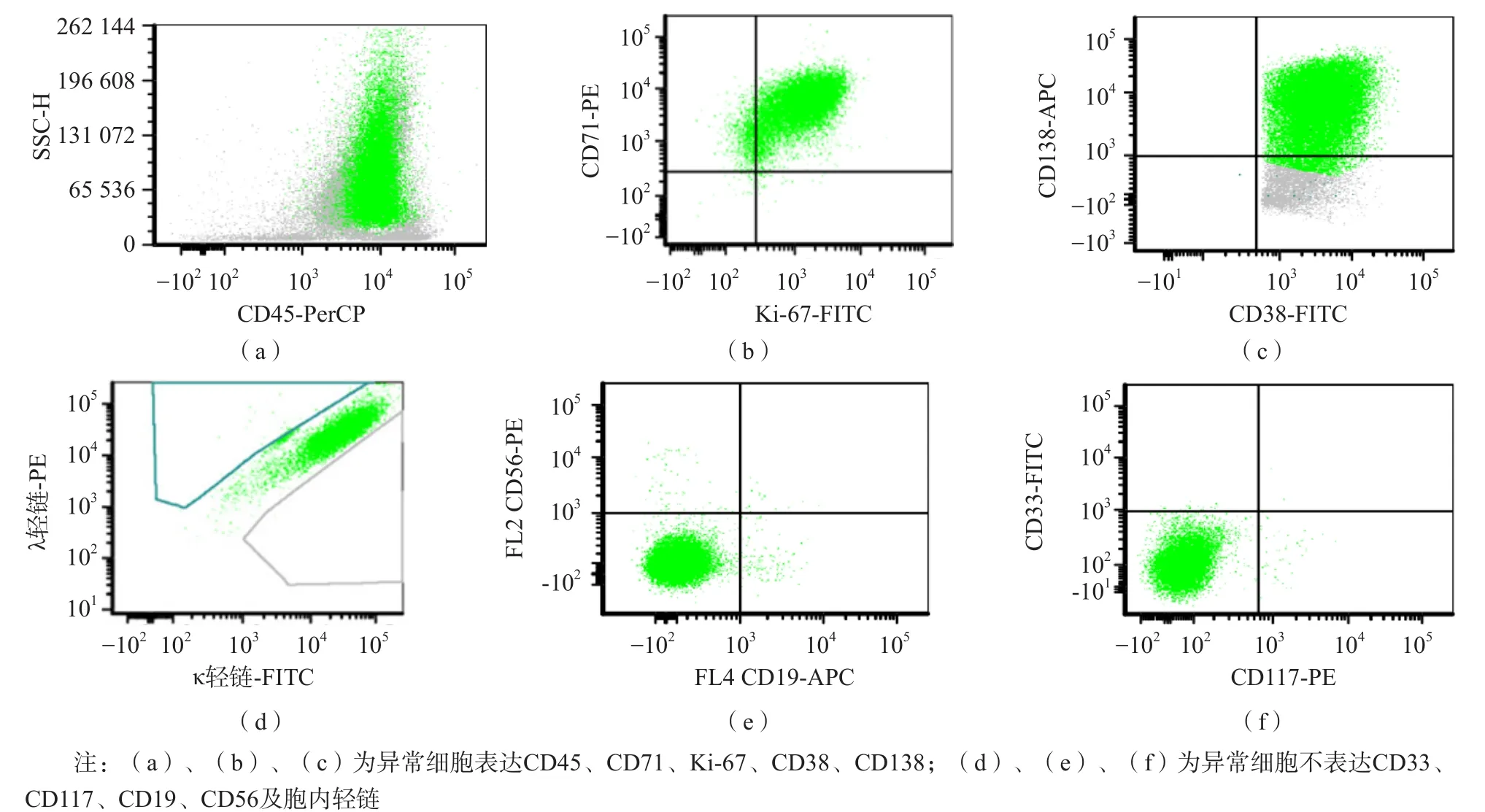
圖2 患者骨髓細胞免疫學表型分析
2 討論
漿細胞骨髓瘤又稱多發性骨髓瘤,是發生在骨髓的多灶性漿細胞惡性腫瘤。其特點是血清中存在單克隆蛋白,溶骨性破壞,病理性骨折,骨痛,有高鈣血癥和貧血等靶器官損傷表現[3]。SAILER等[4]總結了153例病例,認為漿母細胞>30%,可考慮為PPCM,中間可散在有成熟的漿細胞[5]。PPCM又被稱為漿母細胞型漿細胞瘤,是一種較少見的病理類型,最早由BARTL等[6]在1982年提出,將骨髓瘤細胞分為漿母細胞型和漿細胞型。GREIPP等[7]在1984年提出另一種分類標準,將骨髓瘤細胞分為成熟型、中間型、不成熟型和漿母細胞型4種細胞類型。漿母細胞形態學特征為:染色質疏松、胞核直徑 >10 μm或核仁>2 μm、胞質較少、核質比較高。根據此分類,PPCM占骨髓瘤的比例約為8.2%~15.0%[8]。
本病例為女性患者,發病時較為年輕,就診時全身出現多發性的骨質破壞,血清中出現M蛋白,但并無高鈣血癥、貧血、腎功能損傷等表現。骨髓中可見大量異常細胞,占有核細胞的91.6%,該類細胞體積較大,體積為15~30 μm,核染色質疏松,有核仁,胞漿量中等,胞漿深染提示細胞處于較幼稚階段,同時胞漿內存在大量的空泡,在片尾易見異常細胞成團聚集。而PBL的細胞形態學在骨髓涂片中也與PPCM細胞表現相似,但PBL的核染色質并不會表現出經典漿細胞的“clock-face”樣結構,且一般無核周淡染區[9]。APCM的形態特征表現為多形性且多核[10],本例中并未見明顯的多形性。
流式細胞術檢測免疫表型發現,該群異常細胞不表達B淋巴細胞的標志如CD19、CD10、CD20,高比例、高強度表達CD138、CD38。異常細胞表達CD45,CD45作為抗原調制信號的關鍵調節器,可以激活淋巴細胞,在漿細胞早期階段或漿母細胞中可能出現陽性表達[11-12]。在漿細胞分化發育成熟的過程中,CD45 的表達逐漸下降[13]。在PPCM中,多為分化差的漿母細胞,CD45陽性也說明細胞發育幼稚的特征。KIM等[14]發現,CD19-/CDl38+漿細胞可能為骨髓瘤干細胞。CHAIDOS等[15]的研究結果顯示,在免疫缺陷的小鼠模型中移植多發性骨髓瘤患者CDl9+的骨髓瘤細胞都不成功,而CDl9-的骨髓瘤細胞較CDl9+骨髓瘤細胞更具有干細胞特征。所以在出現CD19-的骨髓瘤細胞時要注意在涂片中觀察細胞的分化發育情況,是否為偏早期階段,這對PPCM的診斷有參考價值。而Ki-67為一種核蛋白,表達于Gl、G2、S和M期,而不表達于G0期,因此可反映細胞的增殖狀態[16]。該病例中漿母細胞是一種處于早期階段的漿細胞,增殖活躍,高表達Ki-67和CD71。CD71即轉鐵蛋白受體1,可以反映血液腫瘤細胞的增殖活性,即增殖活躍的腫瘤細胞表面CD71表達水平明顯增高[17]。病理學檢查對于三者的鑒別提供的信息較少,因其均表現為CD38、CD138、MUM1陽性,而CD20、PAX5等為陰性[18]。
由于本例患者就診時已存在多部位的淋巴結腫大,因此與PBL的鑒別就顯得更加重要。PBL的治療原則及手段與PPCM完全不同。PBL是Delecluse在1997年首次報道的發生在免疫缺陷,如獲得性免疫缺陷綜合征患者中的一種較少見的大B細胞淋巴瘤,首發部位常常為口腔、胃腸道等[19],其中有76%感染EB病毒[20]。所以EB病毒的原位熒光雜交檢測對于診斷具有重要價值[21]。PBL的中位發病年齡為58歲,且男性多見[22]。本例患者并無免疫缺陷病史,且EB病毒編碼的小mRNA(Epstein-Barr virus-encoded small RNA,EBER)為陰性,發病年齡又較為年輕,同時伴有骨質破壞,血清免疫球蛋白升高等特點,故診斷為PPCM,腫大的淋巴結考慮為繼發性的骨髓瘤轉移。TADDESSE-HEATH等[23]發現PBL可能與MYC基因的易位有關,而這種易位在多發性骨髓瘤也可見到[24],提示MYC基因可能與B淋巴細胞向漿母細胞轉化過程中異常細胞的形成有關,但對于2種疾病的鑒別作用還有待驗證。其他亞型的彌漫性大B細胞淋巴瘤中Burkitt淋巴瘤細胞胞漿中也會存在大量空泡,但Burkitt淋巴瘤細胞為成熟B細胞表型,表達CD19、CD20、CD10、FMC7、CD22、CD79b、Ki-67,表面輕鏈單克隆表達,根據此特點容易與PPCM區分。APCM是一種少見的、預后非常差的難治性骨髓瘤,在年輕患者中常見,且多為IgA型,易侵犯髓外組織,通常伴有1q21擴增或17p(p53)缺失,t(4;14) 和/或13號染色體異常[25],腫瘤細胞形態表現為多形性,可以在疾病初始期或進展期出現[26]。
綜上所述,檢驗人員需加強對于少見系別淋巴瘤及骨髓瘤的鑒別,診斷需結合血清學檢查、細胞形態、流式細胞術,最重要的是結合臨床病史及表現,并加強對分子生物學、基因測序等檢測手段在疾病診斷及分型中的應用研究。流式細胞術的應用有助于與其他易混淆的淋巴瘤的鑒別,從而提高對于PPCM的及早診斷及診斷準確性,為臨床早期治療提供依據。
