NF-κB在大鼠COPD中的表達及PDTC與肺血管重構的關系
2018-11-06 09:33:52劉曉黎王昌明莫碧文
安徽醫科大學學報 2018年10期
劉曉黎,王昌明,蔣 明,莫碧文
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)是呼吸系統常見病及多發病,特征是持續存在的呼吸道癥狀和氣流受限,原因是氣道和肺泡異常,通常與顯著暴露于毒性顆粒和氣體有關[1],可通過積極預防和治療改善其疾病進程。COPD發病機制并未完全明確,包括:異常炎癥反應、氧化/抗氧化失衡、蛋白/抗蛋白酶失衡等,其中炎癥機制尤為重要。氣道、肺實質以及肺血管的炎癥是COPD的主要病理特征,級聯持續的炎癥反應導致肺血管及氣道重構,肺血管重構是COPD及肺動脈高壓的重要病理基礎。研究[2]表明在COPD早期的異常炎癥反應中致炎因素、炎癥過程及炎癥介質已經開始參與肺動脈重構。核轉錄因子-κB(nuclear factor-κB,NF-κB)是一種調控基因表達的DNA結合蛋白,生物活性廣泛,多項研究證實NF-κB與COPD氣道炎癥、肺血管重構[3]密切相關。實驗通過建立慢支、COPD、COPD并肺動脈高壓動物模型,以NF-κB 特異性抑制劑吡喀烷二硫代氨基甲酸藍(PDTC)作為工具藥,觀測NF-κB表達水平與COPD肺血管重構的關系,探討PDTC在COPD疾病進展中的作用。
1 材料與方法
1.1實驗材料雄性健康SPF級SD大鼠48只,體質量(200 ±20) g,由桂林醫學院動物實驗中心提供,飼養環境為SPF級清潔實驗室;脂多糖(LPS)、PDTC(美國Sigma公司);黃果樹牌烤煙型香煙(貴州中煙工業公司);醫用氮氣、無水氯化鈣及鈉石灰;TRIzol、反轉錄Primescript RT reagent kit試劑盒、PCR SYBR Premix Ex Taq Ⅱ試劑盒(寶生物工程大連有限公司);瓊脂糖(美國Gene Tech公司);NF-κB p65單克隆抗體、羊抗鼠IgG(美國Santa Cruz公司);發光試劑盒(美國Thermo scientific公司);自制有機玻璃艙,BL-420F生物機能實驗系統(成都泰盟科技有限公司);OX-100A數字測氧儀(浙江南加分析儀器廠);TKR-400H電腦小動物呼吸機(南昌新長征醫療科技發展有限公司);IX71倒置熒光顯微鏡(日本Olympus公司);T Personal 48基因擴增儀(德國Biometra公司);Western blot凝膠電泳模具(美國Bio Rad公司);Image-pro-plus6.0圖像分析軟件(美國Media Cybernetics公司);凝膠成像分析系統(上海培清科技有限公司)。
1.2動物模型建立與分組隨機將48只SD大鼠分為8組,實驗組及PDTC藥物干預組各4組,每組6只。制備動物模型,方法如下:實驗組:① 正常對照組:正常飼養28 d后檢測;② 慢支組:第1、14天經氣道內注入LPS 200 μg/次,28 d后檢測;③ COPD組:按照慢支組方法氣道內注射LPS,同時每日香煙煙霧暴露1 h/d,共28 d;④ COPD并肺動脈高壓組:按照COPD組方法,實驗共42 d,最后14 d在香煙煙霧暴露的同時,給予18%低氧8 h/d。PDTC藥物干預組:① 其中慢支干預組、COPD干預組、COPD并肺動脈高壓干預組分別與其相對應的實驗組模型制作方法相同,從第15天起予腹腔內注射PDTC,劑量為100 mg/(kg·d);② PDTC藥物干預空白對照組從第15天起予腹腔內注射與PDTC相等劑量的生理鹽水。
1.3驗證實驗動物模型測定氣道阻力、右心室收縮壓(right ventricular systolic pressure,RVSP)、平均肺動脈壓力(mean pulmonary arterial pressure,mPAP)及觀察肺組織病理改變,物模型制備完成第2天用1%戊巴比妥鈉腹腔注射麻醉大鼠,調節小動物呼吸機,壓力設定為6 kPa,通過頸部氣管插管將大鼠連接于小動物呼吸機,利用壓力傳感器記錄其氣道阻力。后消毒暴露大鼠胸腔,采用肺動脈插管法,將連有壓力換能器的導管插入肺動脈中,生物機能實驗系統通過壓力傳感器記錄肺動脈壓力曲線、RVSP、mPAP。分離大鼠右肺中葉,浸入多聚甲醛固定24 h,距肺門2~3 mm處取材、切片,行HE染色和VG+維多利亞藍染色,觀察肺組織炎癥浸潤情況。
1.4評價肺血管重構指標肺組織切片行維多利亞藍+VG染色,采用倒置熒光顯微鏡及 Image-pro-plus6.0圖像分析系統進行數據測量及分析。觀察肺血管重構相關指標:每例大鼠隨意取5個視野,200倍鏡下記錄直徑<100 μm的肺腺泡內動脈數量并進行分類,計算三型動脈構成比,即肌性動脈(muscular artery,MA)、部分肌性動脈(partially muscular artery,PMA)和非肌性動脈(nonmuscular artery,NMA)。400高倍鏡下計算50 μm<直徑<100 μm肌性動脈的管壁厚度與血管外徑比( the ratio of vascular wall over external diameter,WT%)、管壁面積與血管總面積比(the ratio of vascular wall area over total area,WA%)。右心室肥厚指標:大鼠心臟放血后取出,剪除多余組織,分離右心室游離壁、左心室+室間隔,濾紙反復吸干后稱重,計算右心室肥厚指數(the ratio of the weight of right ventricle/left ventricle and septum,RV/LV+S)值。
1.5檢測肺組織內NF-κBmRNA、蛋白含量用RT-PCR半定量法測定mRNA含量,制備肺組織勻漿,加入TRIzol,提取總RNA,經凝膠電泳鑒定、保存。另取適量RNA測其吸光度值,計算RNA濃度。按照逆轉錄試劑盒操作方法,進行逆轉錄合成cDNA, 保存備用。加入引物序列、模板、DEPC水、設定反應條件后,進行NF-κB PCR擴增反應,再進行瓊脂糖凝膠電泳。然后用凝膠成像分析系統測定每個目的條帶及看家基因β-actin的光密度值,并計算目的基因與β-actin光密度值的比值。
Western blot法檢測肺組織勻漿中NF-κB的表達,于肺組織勻漿中加入蛋白提取液,按照試劑盒步驟提取總蛋白并測定其濃度進行蛋白定量。經12%SDS-PAGE凝膠電泳分離至硝酸纖維素濾膜,5%脫脂奶粉室溫下封閉1 h,加一抗(1 ∶500稀釋)、室溫孵育1 h、PBST洗膜3次加入二抗(1 ∶1 000稀釋)再孵育、洗膜、利用發光試劑盒發光、X線膠片暗室顯影。通過凝膠成像分析系統掃描測定各目的條帶及內參GAPDH的光密度值,計算其比值,代表NF-κB蛋白表達的相對含量。

2 結果
2.1驗證實驗動物模型
2.1.1氣道阻力、mPAP、RVSP比較 與正常對照組比較,慢支組氣道阻力、mPAP、RVSP無明顯變化,COPD組及COPD并肺動脈高壓組氣道阻力、mPAP、RVSP均增高(F=26.329、19.362、13.249,P<0.05);慢支組、COPD組、COPD并肺動脈高壓組氣道阻力依次增高(P<0.05);COPD并肺動脈高壓組RVSP比COPD組增高(P<0.05),但兩組間mPAP差異無統計學意義。PDTC干預后,COPD干預組較COPD組、COPD并肺動脈高壓干預組較COPD肺動脈高壓組mPAP、RVSP明顯下降(t=2.673、3.864、2.712、3.841,P<0.05)。見表1。
2.1.2肺組織病理形態學改變 肺組織切片行HE、VG+維多利亞藍染色染色后觀察,相比正常對照組,慢支組小氣道管壁可見炎癥細胞浸潤,支氣管纖毛柱狀上皮層見杯狀細胞增生,管腔內炎細胞聚集及黏液分泌增多,平滑肌層未見增厚或斷裂。COPD組氣道腔內炎細胞聚集及黏液蓄積較慢支組更為明顯,管壁內浸潤大量炎癥細胞,柱狀上皮杯狀細胞增生明顯,平滑肌層增厚,部分肌層已經斷裂,黏膜下及外膜見膠原纖維增生,同時有肺氣腫形成,部分可見肺泡壁斷裂形成肺大泡。VG+維多利亞藍染色可見紅染的膠原沉積多于正常對照組。COPD并肺動脈高壓組病理組織學改變與COPD組相似,但氣道管腔內及管壁炎癥細胞浸潤程度稍輕,平滑肌層增厚更為明顯。經PDTC干預后, COPD干預組氣道管壁炎癥細胞浸潤較COPD組減輕。COPD并肺動脈高壓干預組炎癥浸潤,氣道平滑肌層斷裂、肺泡壁斷裂程度較COPD并肺動脈高壓組減輕。見圖1、2。
2.2動脈構成比、肌性動脈WT%和WA%、RV/LV+S比較相比正常對照組,慢支組肺血管重構指標無明顯改變,COPD組肌化型動脈百分比升高(F=2.663,P<0.05),對直徑在50~100 μm的肌化型動脈進行圖像分析WA%、WT%比值無升高(F=22.53、28.856,P>0.05);COPD并肺動脈高壓組肌化型動脈百分比升高,WA%、WT%比值亦升高(P<0.05)。相比各實驗組,COPD干預組、COPD并肺動脈高壓干預組肌化型動脈百分比下降(t=5.285、8.179,P<0.05)。COPD并肺動脈高壓干預組較COPD并肺動脈高壓組WA%、WT%比值亦有所下降(t=2.852、3.65,P<0.05)。見表2、圖2。 相比正常對照組,慢支組RV/LV+S無變化;COPD組、COPD并肺動脈高壓組RV/LV+S值增高(F=3.995,P<0.05),上述兩組間差異無統計學意義。PDTC干預后,COPD并肺動脈高壓干預組RV/LV+S較COPD并肺動脈高壓組降低(t=2.663,P<0.05)。見表2。
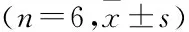
表1 各組氣道阻力、RVSP、mPAP比較
與對應實驗組比較:*P<0.05;與正常對照組比較:#P<0.05

圖1 肺組織病理學變化 HE×20
A:正常對照組;B:慢支組;C:COPD組;D:COPD并肺動脈高壓組;E:干預空白對照組;F:慢支干預組;G:COPD干預組;H:COPD并肺動脈高壓干預組

圖2 肺小動脈結構變化 VG+維多利亞藍×20
A:正常對照組;B:慢支組;C:COPD組;D:COPD并肺動脈高壓組;E:干預空白對照組;F:慢支干預組;G:COPD干預組;H:COPD并肺動脈高壓干預組
表2 肺血管重構相關指標、RV/LV+S分析
與對應實驗組比較:*P<0.05;與正常對照組比較:#P<0.05;與COPD并肺動脈高壓組比較:▽P<0.05

圖3 肺組織NF-κB mRNA RT-PCR電泳圖
M:Marker; A:正常對照組;B:慢支組;C:COPD組;D:COPD并肺動脈高壓組;E:慢支干預組;F:COPD干預組;G:COPD并肺動脈高壓干預組
2.3大鼠肺組織內NF-κBmRNA含量及蛋白含量
2.3.1肺組織NF-κB mRNA相對含量 相比正常對照組(0.187±0.054),慢支組(0.240±0.054)NF-κB mRNA表達無明顯變化(t=-1.739,P>0.05),COPD組(0.331±0.048)較正常對照組(0.153±0.061)、COPD并肺動脈高壓組(0.392±0.068較正常對照組(0.144±0.060)NF-κB mRNA表達顯著增強(t=-5.613、-6.633,P<0.05);同時COPD并肺動脈高壓組(0.646±0.075)NF-κB mRNA表達明顯強于COPD組(0.541±0.058)(F=70.779,P<0.05)。COPD干預組(0.243±0.051)較COPD組(0.331±0.048)、COPD并肺動脈高壓干預組(0.288±0.069)較COPD并肺動脈高壓組(0.392±0.068)NF-κB mRNA表達明顯減弱(t=3.025、3.037,P<0.05)。見圖3。
2.3.2肺組織NF-κB蛋白相對含量 相比正常對照組(0.832±0.202),慢支組(0.822±0.208)NF-κB蛋白含量無顯著變化,COPD組(1.657±0.230)較正常對照組(0.511±0.116)、COPD并肺動脈高壓組(1.697±0.233)較正常對照組(0.517±0.118)NF-κB蛋白表達增強(t=-10.898、-11.062,P<0.05)。慢支組(0.376±0.117)、COPD組(0.801±0.177)、COPD并肺動脈高壓組(1.550±0.203)NF-κB蛋白表達水平依次遞增(F=73.857,P<0.05)。COPD干預組(0.874±0.147)、COPD并肺動脈高壓干預組(0.895±0.190)NF-κB蛋白表達均較其實驗組(1.657±0.230)、(1.697±0.233)減弱(t=17.795、5.949,P<0.05)。見圖4。
3 討論
感染、吸煙是COPD的兩大主要致病因素。研究[4]顯示慢性炎癥、低氧因素與COPD的肺血管重構密切相關,最終導致肺動脈高壓、慢性肺源性心臟病。本實驗制備COPD各階段動物模型,結果顯示慢支組無氣道阻力增高,病理切片見氣道纖毛倒伏、黏連,杯狀細胞增生,氣管腔內炎細胞聚集和分泌物增多;COPD組氣道阻力增高,管腔內炎細胞聚集及黏液蓄積,管壁內大量炎癥細胞浸潤,柱狀上皮杯狀細胞增生明顯,平滑肌層增厚,黏膜下及外膜膠原纖維增生,部分肌層斷裂,肺氣腫形成,部分肺泡壁斷裂形成肺大泡,mPAP、RVSP升高,RV/LV+S、肌化型動脈百分比增高;COPD并肺動脈高壓組病理改變與COPD組相似,但氣道管腔內及管壁炎癥浸潤程度稍輕,平滑肌層增厚更明顯。上述模型病理學改變均與人類慢支、COPD的病理特征吻合,COPD組與COPD并肺動脈高壓組分別代表COPD肺血管重構的不同階段,COPD組代表早期階段,表現為肌化型動脈百分比增多;COPD并肺動脈高壓組代表肺血管重構進展,腺泡內肌化型動脈百分比增高,管壁厚度增加,WA%、WT%比值增高。
NF-κB屬于Rel蛋白家族,廣泛存在于真核細胞內,生物學功能包括參與炎癥、免疫反應、氧化應激、細胞增殖及凋亡、自身的轉錄、細胞周期的調控、腫瘤的發生[5-6]等。NF-κB在氣道炎癥、血管重塑過程中至關重要[7-8],是氣道氧化應激的重要標志物之一,影響IL-1β、iNOS、COX-2、G-CSF、MCP-1等[9]炎癥介質基因表達和釋放,并通過反饋環路導致炎癥過程的放大和持續。上述炎癥介質可影響VFGF、TGF-β、FGF等的調節,進一步影響肺血管重構。本實驗通過建立COPD相關動物模型,RT-PCR、Western blot檢測顯示COPD組、COPD并肺動脈高壓組肺組織內NF-κB mRNA表達逐漸增強、蛋白含量逐漸增高;且NF-κB mRNA、蛋白的表達與肺血管重構相關指標變化一致。提示NF-κB表達與大鼠COPD疾病的發展關系密切。
PDTC是一種新型抗氧化劑,為NF-κB特異性抑制劑,主要通過泛素化蛋白酶降解途徑影響NF-κB的核移位來抑制其活性[10-11],PDTC能有效阻斷NF-κB下游低氧誘導因子的激活,延緩COPD疾病的進展。章忠渭 等[12]發現PDTC能抑制重癥急性胰腺炎大鼠胰腺NF-κB活化。Kan et al[13]等研究表明PDTC 可影響NF-κB、Fas、TNF-α等因子的活性,改善肺損傷。本實驗借助PDTC為工具藥,檢測PDTC藥物干預組NF-κB mRNA表達及蛋白含量均較對應實驗組減少、減弱,RVSP、mPAP值降低,COPD并肺動脈高壓干預組RV/LV+S值降低,肌化型動脈WT%、WA%比值下降,提示肺血管重構得到不同程度的改善。
綜上所述,NF-κB的表達與大鼠COPD疾病的發展關系密切,在肺血管重構中有相當重要的作用,早期利用PDTC干預NF-κB表達可抑制COPD肺血管重構。與COPD階段不同的是,在慢支模型組中未檢測到肺組織NF-κB異常表達,究其原因,可能與檢測時間為第二次氣道內注入脂多糖2周之后,以及機體的負反饋調節有關。NF-κB在COPD發病過程中有不可忽視的作用,NF-κB抑制劑PDTC可在一定程度上抑制COPD肺血管重構,延緩COPD疾病的進程,其聯系有可能成為COPD疾病新型抗炎類藥物的作用靶點,為臨床治療開拓新方向,但其具體機制仍有待進一步研究及探討。
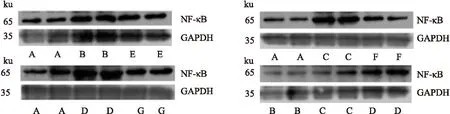
圖4 肺組織NF-κB Western blot電泳圖
A:正常對照組;B:慢支組;C:COPD組;D:COPD并肺動脈高壓組;E:慢支干預組;F:COPD干預組;G:COPD并肺動脈高壓干預組
