基因編輯技術:進展與挑戰
2018-11-22 01:32:48盧俊南潘燕平陳映羲戴俊彪
中國科學院院刊 2018年11期
盧俊南 褚 鑫 潘燕平 陳映羲 溫 欒 戴俊彪*
1 中國科學院深圳先進技術研究院 合成生物學研究所 合成基因組學研究中心 深圳市合成基因組學重點實驗室深圳 518055
2 中國科學院 重大科技任務局 北京 100864
3 中國科學院深圳先進技術研究院 合成生物學研究所 定量合成生物學研究中心 深圳 518055
生命科學的迅速發展使得我們從生物遺傳信息的“讀取”階段進入到后基因組時代,基因組的“改寫”乃至“全新設計”正逐漸成為現實。以設計創造新生命體為目標的合成生物學在此背景下迅速發展,并在醫藥、制造、能源等領域顯現了巨大的應用前景。基因組的從頭合成和針對天然基因組的規模改造分屬合成基因組學和基因編輯領域,均為當前合成生物學研究的熱點。合成基因組學涉及基因組的從頭設計、構建和功能表征等,屬于自下而上的生物學研究策略;而基因編輯側重于通過對現有基因組進行刪除、替換、插入等分子操作,進而改寫遺傳信息,屬于自上而下的生物學研究策略。以上兩者的有機結合將極大地推動生物制造、疾病治療等領域的革新;同時二者也將為后基因組時代,功能基因組學的研究提供強有力的技術手段。新生命體系的從頭設計與合成不僅需要基因組序列的合成、拼接及轉移等技術,也需要高效率、低脫靶率的編輯技術以實現在基因組上進行大規模的編輯改造。在不斷的探索研究中,基因編輯技術已經從最初依賴細胞自然發生的同源重組,發展到幾乎可在任意位點進行的靶向切割,其操作的簡易和高效極大地推動了物種遺傳改造的發展。基因編輯可為合成生命的進一步改造提供手段,為新物種的創造提供更多的可能性。
1 基因編輯的原理
基因編輯技術是對生物體 DNA 斷裂的現象及其修復機制的應用。作為一種常見的分子生物學事件,在分裂活躍的哺乳動物細胞中,DNA 雙鏈斷裂(DNA doublestrand breaks,DSBs)每天會發生[1-4]。DSBs 發生后細胞可以通過多種方式進行修復,包括經典的非同源末端修復(non-homologous end joining,NHEJ),選擇性末端修復(alternative end joining,a-EJ),單鏈退火修復(single-strand annealing,SSA)和同源重組修復(homologous recombination,HR)[4]。HR 可以進行精確無誤的修復,但是需要同源模板的存在[5-7];NHEJ 則是將很大程度上沒有同源性的兩個 DNA 末端直接連接實現修復[7],此過程中,兩個末端在大多數情況下都會發生若干核苷酸的缺失,是一種不精確的修復機制[8]。而作為輔助性的修復機制,a-EJ 和 SSA 均需要更大幅度的末端單鏈切除[4],這也會導致遺傳信息的丟失。基于 DNA 斷裂修復的原理,如果在細胞中人為提供特定的同源重組模板,待目標 DNA 自然發生或者人為誘導產生 DSBs,觸發同源重組修復,就有機會把特定 DNA 序列進行刪除或者插入外源基因。在不提供同源模板的情況下,利用 NHEJ、a-EJ 及 SSA 的不精確修復機制可以實現基因的突變和敲除。傳統的基因編輯借助細胞內自然發生的 DSBs 實現靶向整合[9],達到基因敲除、替換等目的。然而,在真核生物細胞里面,通過自發雙鏈斷裂實現目的基因編輯的概率通常低至百萬分之一[10,11]。人為使用化學誘導劑或者輻射處理等方法,或者使用轉座子技術也可以實現基因的突變,但是這些突變是隨機的,需要后續進行大量的篩選工作來獲得所需的基因型。定點基因編輯技術是進行基因功能研究和物種定向改造的優選策略。
2 重組核酸酶介導的基因編輯技術
2.1 鋅指核酸酶技術
人工核酸酶技術的發展使人為定點誘導DSBs成為現實,其中鋅指核酸酶技術(zinc finger nucleases,ZFNs)就是一個里程碑式的突破,也稱為第一代基因編輯技術。ZFN 由鋅指蛋白(zinc finger protein,ZFP)和 FokI 內切酶的核酸酶結構域組成,前者負責識別,后者負責切割 DNA。ZFP 是自然存在的蛋白結構,其由鋅指結構(zinc finger,ZF)組成,ZF 能識別特定的 3 個連續堿基對[11](圖 1a),因此可通過串聯 ZF 的數量調整 ZFN 的識別特異性。FokI 通過 N 端與 ZFP 連接,由于 FokI 以二聚體的形式發揮切割作用,ZFN 使用時需要成對設計[12]。作為新型基因編輯工具,ZFN 從 2001 年開始被陸續用于不同物種的基因編輯[11,13]。
2.2 TALENs 技術
ZFN 技術將基因編輯引領進了不再單純依賴自然發生 DSBs 的時代,但其存在很大的局限性,如成本高、難以實現多靶點編輯等[11]。而 TALE(transcription activator like effector)基序的發現催生了第二代基因編輯技術——TALENs(TALE nucleases)。TALEN 的構造與 ZFN 類似,由 TALE 基序串聯成決定靶向性的 DNA 識別模塊,與 FokI 結構域連接而成。與 ZF 基序不同,一個 TALE 基序識別一個堿基對(圖 1b),因此串聯的 TALE 基序與所識別的堿基對是一一對應的關系[11]。研究發現,對于相同的靶點 TALENs 有與 ZFNs 相同的切割效率,但是毒性通常比 ZFNs 的低,另外其構建也比 ZFNs 容易[11]。然而,TALENs 在尺寸上要比 ZFNs 大得多,而且有更多的重復序列,其編碼基因在大腸桿菌中組裝更加困難[11]。
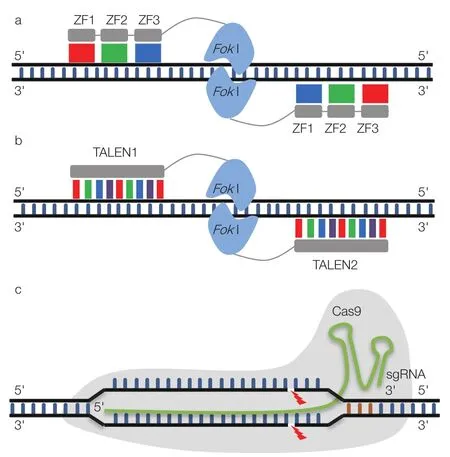
圖1 ZFN、TALEN 及 CRISPR/Cas9 基因編輯技術
3 RNA引導的基因編輯技術
3.1 CRISPR/Cas9 技術
CRISPR/Cas 系統原本是細菌和古菌進化出來用于抵御外來病毒及質粒 DNA 的適應性免疫系統。II 型CRISPR/Cas 系統依賴于外源 DNA 片段在規律成簇的短間隔回文重復(clustered regularly interspaced short palindromic repeat,CRISPR)位點整合,其經過轉錄及剪切后產生短的 CRISPR RNAs(crRNAs),crRNA 與反式轉錄的 crRNA(trans-activating crRNA,tracrRNA)退火結合,然后引導 Cas9(CRISPR associated protein 9,Cas9)蛋白介導序列特異性的外源 DNA 降解[11,14-16]。Jinek 等[17]發現 Cas9 發揮靶向切割作用所依賴的 crRNA 和 tracrRNA 可以融合為一體,作為 sgRNA(single guide RNA)(圖 1c)。隨后,若干研究組陸續報道 CRISPR/Cas9 系統可用于人類細胞的靶向基因編輯[18-21]。與 ZFNs 和 TALENs 技術相比,CRISPR/Cas9 的設計要簡單得多,而且成本很低,對于相同的靶點,CRISPR/Cas9 有相當甚至更好的靶向效率[11]。
3.2 CRISPR/Cas9 衍生技術
隨著 CRISPR/Cas 系統的研究不斷深入,Cas9 核酸酶的催化機制也被揭示[17],并且可以通過特定位點氨基酸的突變獲得單鏈靶向剪切功能的 Cas9 切刻酶(Cas9 nickase,Cas9n)或者全失活的 Cas9(dead Cas9,dCas9),而這些不同的 Cas9 核酸酶,衍生出了適用范圍更為廣泛的基因編輯系統。
(1)CRISPR/Cas9-nickase 基因編輯技術。CRISPR/Cas9 系統使用的 crRNA 能容受一定程度的錯配導致脫靶效應的產生,限制了 CRISPR/Cas9 系統在高精度編輯中的應用[22]。為了提高 Cas9 編輯系統的精度,Ran 等[22]巧妙利用 Cas9 D10A 突變體具備切刻酶活性的特性,設計了“一個位點,雙 sgRNA 靶向”的策略,即 CRISPR/Cas9-nickase 基因編輯技術(圖 2a)。其原理與 ZFN 和 TALEN 類似,兩個 Cas9n/sgRNA 復合物同時靶向一個位點,分別切割其中一條 DNA 鏈,實現雙鏈斷裂,誘導 NHEJ 或者 HR 修復。使用這種策略,細胞系中基因編輯的脫靶效率最高可以降低近 4 個數量級。
(2)CRISPR/dCas9-FoKI基因編輯技術。同樣是為了解決 CRISPR/Cas9 技術的脫靶問題,Guilinger 等[23]采用了基于 dCas9 的策略。理論上 dCas9/sgRNA 只能起到單純的靶向引導作用,無法誘導 DNA 的斷裂,類似于 ZFN 或者 TALEN 中的 DNA 結合結構域。為了實現 DNA 的切割,其引入的 FoKI 核酸內切酶的切割結構域(圖 2b),與 dCas9 連接做成融合蛋白 fCas9,這與 ZFN 及 TALEN 的設計策略如出一轍。在人類細胞的基因編輯中,fCas9 的特異性比野生型 Cas9 要高出 140 倍以上。而在高度類似的脫靶位點上,fCas9 的特異性要比 Cas9n 至少高出 4 倍。fCas9 的應用將進一步豐富 Cas9 工具箱,提供更完善的基因編輯工具。
(3)基于 CRISPR/dCas9 的單堿基編輯技術。人類大多數的遺傳病與基因的點突變有關[24],如果能通過精確的手段進行修復,可能會帶來新的治療策略。在提供同源重組模板的情況下,定點突變可以通過 CRISPR/Cas9 來實現,但是其誘導的 NHEJ 修復可能帶來的堿基隨機插入、缺失是潛在的危險因素。Cas9 的突變體 Cas9n、dCas9 無切割雙鏈 DNA 的功能,但可以發揮尋靶定位作用;如果有能催化特定堿基轉換的蛋白/結構域可用,則可參考 CRISPR/dCas9-FoKI 蛋的設計,構建 CRISPR/Cas9n/dCas9 導向的單堿基編輯技術。Liu 課題組將源自大鼠的胞嘧啶脫氨酶(APOBEC1)與 dCas9 融合(圖 2c),發現其可以定點將 C 轉變為 U,然后在后續的 DNA 復制或者修復作用下實現 C : G 堿基對到 T : A 的轉變[24]。隨后日本神戶大學的 Kondo 課題組[25]及我國上海交通大學的常興課題組[26]也報道了類似研究成果。為了實現 A : T 到 G : C 的轉換,Liu 課題組采用蛋白質進化工程手段對大腸桿菌的 tRNA 腺苷脫氨酶(TadA)進行改造,他們獲得的第 7 代腺嘌呤堿基編輯器(adenine base editors,ABEs)能高效介導 A : T 堿基對到 G : C 的轉變[27]。即,C 到 T 以及 G 到 A 堿基之間的自由轉換已經實現,將來有可能做到 4 種堿基的任意轉換。
(4)基于 CRISPR/dCas9 的基因表達調控技術。除了直接對 DNA 進行編輯,CRISPR/Cas 系統在基因表達調控上也可發揮作用[28,29]。例如,利用 dCas9 無核酸內切酶活性但仍能與 DNA 結合的特點,可直接阻礙其結合 DNA 與其他因子的結合,影響基因的表達(圖 2d)。如果將轉錄抑制因子或者激活因子與 dCas9 融合,則可以實現靶基因的抑制與激活,為基因功能研究提供靈活的操作手段。
3.3 CRISPR/Cas12a 基因編輯技術
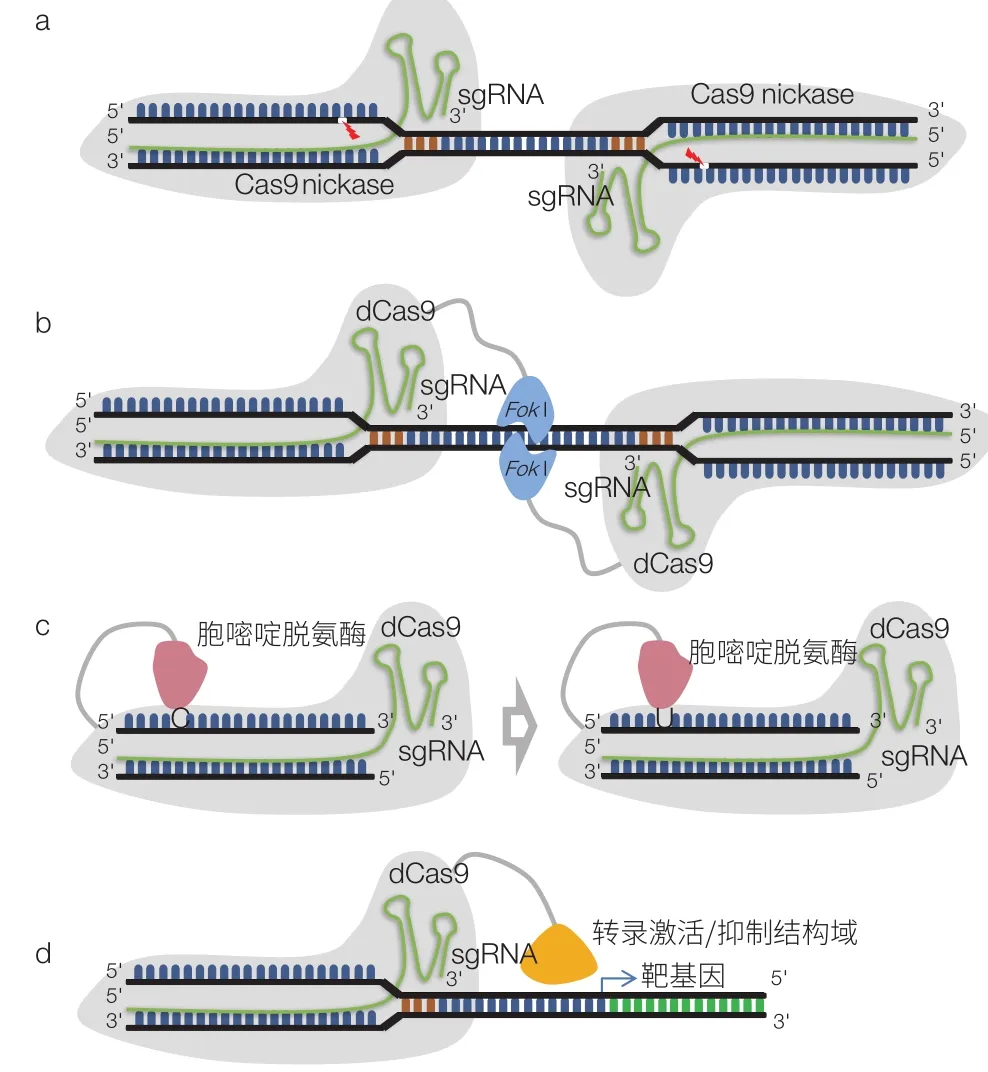
圖2 系列CRISPR/Cas9衍生基因編輯技術
CRISPR/Cas9 技術受富含 G 堿基的 PAM 序列限制,不能實現任意序列的靶向。同時 Cas9 蛋白分子量過大,某些情況下不便使用。實際上幾乎所有的古細菌和眾多的細菌都采用 CRISPR/Cas 機制進行免疫防御[30],其中包含多種 CRISPR/Cas 系統。已經表征過的Ⅱ類 CRISPR/Cas 系統,均屬于采用 Cas9 家族核酸酶作為效應因子的Ⅱ型 CRISPR/Cas 系統,而在普氏菌和弗朗西斯氏菌屬(Prevotella 和 Francisella 1)中存在另一個Ⅱ類 CRISPR/Cas 系統,其被歸為Ⅴ型 CRISPR/Cas 系統[30]。2015 年,張鋒團隊報道 V 型系統中的 Cpf1(CRISPR from Prevotella and Francisella 1)(現稱“Cas12a”)是有功能的細菌免疫機制并能在人類細胞中介導有效的基因編輯[30]。CRISPR/Cas12a 具有 CRISPR/Cas9 沒有的優點[30],其中之一就是 Cas12a 需要的是富含 T 堿基的 PAM 序列,有助于其在基因組富含 A/T 堿基的物種中使用。上海科技大學的陳佳課題組將無 DNA 切割活性的 dCas12a 與大鼠源的胞嘧啶脫氨酶(APOBEC1)融合,發現其與基于 Cas9 的堿基編輯器類似,能有效地催化人類細胞中 C 到 T 堿基的轉換[31]。由于識別的是富含 T 堿基的 PAM 序列,基于 Cas12a 的堿基編輯系統能與基于 Cas9 的堿基編輯系統互補,為相關基礎研究及將來的臨床應用提供更全面的技術條件。
3.4 基于 CRISPR/Cas 系統的RNA編輯技術
除了對 DNA 進行編輯,張鋒課題組發現 CRISPR 蛋白家族的 C2c2(現稱 Cas13a)可以靶向切割 RNA[32],隨后他們證實 Cas13a 可在哺乳動物細胞中靶向降低 RNA 的水平[33]。利用 Cas13a 的 RNA 靶向功能,CRISPR/Cas13a系統被開發為 RNA 檢測器用于疾病診斷[34]。張鋒團隊后續又發現了 Cas13b[35],其同樣具有 RNA 靶向和編輯功能。另外,Cas13c 和 Cas13d 也具有類似功能[36]。RNA 編輯技術的建立,進一步拓展了 CRISPR/Cas 基因編輯技術的應用范圍。
4 DNA介導的基因編輯工具
根據堿基互補配對原則,及引物設計原理,Oligo DNA 理論上也可以用于引導核酸內切酶靶向切割雙鏈 DNA。類似于 ZFN 和 TALEN,可用 Oligo DNA 替代其 DNA 結合結構域,關鍵是需要找到 Oligo DNA 與切割結構域 FoKI 的合適連接方式。或者是尋找類似于 Cas9 的天然蛋白,本身具備核酸內切酶功能,同時能結合一定長度的引導 Oligo DNA。由于 DNA 本身有更好的穩定性,Oligo DNA 制備成本低,可以簡化基因編輯的操作程序,Oligo DNA 引導的基因編輯工具有其他編輯工具不可比擬的優點,值得深入研究開發。特別是當前主流基因編輯工具全為國外專利,對今后國內基因編輯相關產品的上市不利,具備自主知識產權的基因編輯技術亟待開發。
2016 年 9 月,Genome Biology 刊登了我國南京大學研究團隊研發的新型基因編輯工具:結構引導的核酸內切酶(structure-guided endonuclease,SGN)[37]。SGN 是典型的 Oligo DNA 引導基因編輯工具,本質上也是重組核酸酶的設計與應用。其 N 端是能識別 DNA 3′ 末端翹翼(3′ flap)的核酸內切酶(flap endonuclease-1,FEN-1),C 端則是 FoKI 核酸內切酶的 DNA 切割結構域(Fn1)。SGN 通過識別引導 DNA(guide DNA,gDNA)與特點靶點結合產生的 3′ flap 進行定位切割。不同于 CRISPR/Cas9 及其系列衍生技術,SGN 沒有 PAM 限制,理論上可以靶向任何序列。實驗結果顯示 SGN 的切割活性不依賴于靶點的序列,其可用于斑馬魚基因編輯但是效率尚有很大的提升空間。SGN 作為我國科學家的原創性研究結果,其優點無疑顯著,如:gDNA 非常容易獲得并且可根據需要精確控制其用量;可以通過調整 gDNA 的長度將錯配的可能降到最低等。
5 我國基因編輯技術發展面臨的挑戰與機遇
近年來,我國在基因編輯技術方面取得了矚目的進展。但是我們要清醒地認識到,目前基因編輯,尤其是 CRISPR/Cas9 相關的核心專利基本都是掌握在其他國家手中。未來基因編輯技術及基因編輯的細胞制品等走向臨床應用,以及基因編輯農作物走向市場所產生的巨大利潤都會因此而受到重大損失。開發具備自主知識產權的核心技術才有可能在這場生物技術革命中獲得發展,包括對現有基因編輯技術的缺陷進行修正,以及通過技術組合等,搶先獲得現有基因編輯技術的改進版本和增強版。同時也借助生物信息學手段挖掘潛在的 CRISPR 核酸酶和 DNA 引導的核酸酶等,開發新型基因編輯技術。此外,基因編輯技術,尤其是 CRISPR/Cas 技術已經廣泛用于各物種的基因編輯,除了常見的模式生物,如線蟲、果蠅、斑馬魚、小鼠等,還有豬、狗、猴等大型動物,以及水稻、小麥等常見農作物。將現有基因編輯技術拓展到其他生物也是新的突破,尤其對于我國特有的生物資源,其過程往往涉及技術的調整和改進,一方面可以產生新的技術,另一方面可為目標生物的功能基因組研究以及遺傳改良提供有效手段。
目前,CRISPR/Cas9 等基因編輯技術已經顯示巨大的應用價值,但是其在編輯效率、精確度及脫靶效應等方面,以及其走向臨床應用等尚有很多問題需要解決。病毒載體可應用于人類基因治療,但是其裝載容量有限,常規的 Cas9 蛋白過大,不利于使用。一方面可以從自然界尋找更小的 Cas9 蛋白,另一方面可以通過基因工程手段進行適當的刪減。而在減少脫靶效應方面,除了上述提到的 CRISPR/Cas9-nickase 和 CRISPR/dCas9-FoKI基因編輯技術,還可以對 Cas9 蛋白本身進行定點突變等來增強其特異性。這些工作充滿著挑戰性,但也是我們發展的機遇和突破口。
近年來,我國科學家在基因編輯領域取得了長足進展。上述提及常興課題組和陳佳課題組在堿基編輯技術上的突破,此外,哈爾濱工業大學黃志偉課題組和中國科學院生物物理研究所王艷麗課題組在 Cas/sgRNA 復合物的結構解釋上也作出了突出貢獻,可為基因編輯原理的理解提供重要參考。中國科學院遺傳與發育生物學研究所高彩霞課題組等則在植物基因編輯上取得重要進展。中國科學院廣州生物醫藥與健康研究院賴良學課題組、昆明理工大學季維智課題組、中國科學院神經科學研究所楊輝課題組等,利用基因編輯技術成功獲得多種疾病動物模型。我國在新型基因編輯技術開發上也取得了一定突破,DNA 引導的基因編輯技術有著獨特的優勢,值得加大投入以解決編輯效率低等問題。
此外,合成生物學作為新近迅速發展的交叉學科,已在生物醫藥、能源、新材料等領域展現越來越廣泛的應用潛力。基因組合成和基因編輯,涉及的操作廣度、深度不同,技術體系也不同。但其本質上都是通過遺傳改造,獲得具有特定功能的生命體,服務科研與生產,二者有機融合是水到渠成的趨勢。比如,在合成的基因組中可以引入基因編輯體系,為新生物體進一步改造及應用提供更多的可能性。SCRaMbLE(synthetic chromosome rearrangement and modification by loxP-mediated evolution)[38]是一種快速的染色體重排修飾技術,可以加速生物體的進化,其本質上是一種非定點的誘導型基因編輯技術。生物的進化往往需要漫長的歷史過程,難以快速獲得具備某類性狀的物種。合成生物學可以通過基因組設計,引入特定的功能模塊,以獲得目標工程細胞;然而,基于目前的成本和技術限制,盡管可以有很多候選基因組設計,想要獲得一個大容量合成細胞庫并從中篩選獲得最優設計的可能性不大。而 SCRaMbLE 是一種潛在的高性價比手段。Sc2.0 計劃擬在合成的酵母基因組中插入約 5 000 個 loxPysm 位點[39]。該計劃近期發表的系列成果[40-46]表明,SCRaMbLE 系統可以誘導獲得增強型工業用酵母菌株,提目標代謝物的產量等。
當前,首個全人工合成的真核生物——Sc2.0 已經接近完成,更高等生物的全基因組合成也已經提上日程[47]。然而,超大基因組的合成尚有很多挑戰。通過基因編輯對現有物種進行局部基因組改造實際為傳統基因工程的升級,其不大可能實現全面的基因組改寫。就目前而言,基因組合成與基因組編輯的結合將是一個十分具有潛力的研究方向,可解決目前超大基因組從頭合成面臨的各項困難,為合成基因組學在超大基因組物種基礎研究及物種改造中的應用提供可能。這也是我國在合成生物學領域及基因編輯領域可以把握的契機。
