spo0A基因缺失對克勞氏芽孢桿菌發酵生產性能的影響
2018-12-07 07:32:24原梨萍王瑞明路福平汪俊卿
中國釀造 2018年11期
原梨萍,肖 靜,王瑞明,路福平*,汪俊卿,李 玉
(1.天津科技大學 生物工程學院 工業微生物教育部重點實驗室,天津 300457;2.齊魯工業大學 生物工程學院 山東省微生物酶技術重點實驗室,山東 濟南 250353)
克勞氏芽孢桿菌(Bacillus clausii)是一類好氧型、內生抗逆孢子的革蘭氏陽性菌,具有很好的耐受性,能在極端環境下生長。近年來,芽孢形成及萌發過程中關鍵基因的研究[1-2]備受青睞,而克勞氏芽孢桿菌由于其生理特性及基因信息不明確,使其在芽孢方面的研究及工業上的應用較為落后。目前,國內外科研人員以模式微生物枯草芽孢桿菌為主對芽孢形成過程及關鍵基因的調控做了大量深入的研究[3-7],其中,以Spo0A、sigma因子及五種磷酸激酶基因的研究居多[8-13]。芽孢的生成是一個多層次分階段的過程,其中Spo0A是細胞從營養生長期進入芽孢形成時期的關鍵應答調節蛋白(responseregulatorprotein,RSP),Spo0A-PO4通過結合到基因“OA-box”序列上可調節500多種基因的表達。
目前國內外研究發現,spo0A基因缺失對枯草芽孢桿菌(Bacillus subtilis)、梭狀芽孢桿菌、蘇云金芽孢桿菌(Bacillusthuringiensis)等多種芽孢桿菌的芽孢生成均產生有效的抑制作用[14-16],而且該基因一定程度上影響菌體生物量及代謝產物的產量,SANDOVAL NR等[17]敲除梭狀芽孢桿菌spo0A基因后不僅對芽孢生成產生影響,而且有效提高了菌體生物量及丁醇產量。目前國內外關于spo0A基因缺失影響淀粉酶產量的報道少見。本實驗通過基因敲除技術構建spo0A基因缺失菌株,研究發現spo0A基因的缺失對菌體生物量、芽孢生成率及淀粉酶活力均產生影響,該基因在菌種改良上可以作為提高淀粉酶產量的靶點,對后續芽孢缺失型工業生產菌株的構建具有重要意義,為酶工業遺傳育種提供了新的選擇,也為高產淀粉酶芽孢桿菌工程菌株的構建奠定了基礎。
1 材料與方法
1.1 材料與試劑
1.1.1 菌株及質粒
克勞氏芽孢桿菌(Bacillus clausii)QL-1、含pHT01質粒的大腸桿菌(Escherichia coli)JM109:山東省微生物工程重點實驗室保存。
1.1.2 試劑
限制性核酸內切酶:大連TakaRa公司;氯霉素、Ezup柱式基因組脫氧核糖核酸(deoxyribonucleicacid,DNA)抽提試劑盒、SanPrep柱式DNA膠回收試劑盒:生工生物工程(上海)有限公司;高純度質粒小量快速抽提試劑盒:北京艾德萊生物科技有限公司;其他試劑均屬于國產分析純。引物由生工生物工程(上海)有限公司合成。
1.1.3 培養基
LB培養基:酵母浸粉5g/L、蛋白胨10g/L、氯化鈉10g/L、pH 7.0。
可溶性淀粉培養基:牛肉膏5 g/L、蛋白胨10 g/L、氯化鈉 5 g/L、可溶性淀粉 2 g/L、pH 7.0~7.2、瓊脂 20 g/L。
菌體增殖培養基:酵母浸粉5 g/L、蛋白胨10 g/L、氯化鈉10 g/L、山梨醇91 g/L。
菌體復蘇培養基:酵母浸粉5 g/L、蛋白胨10 g/L、氯化鈉10 g/L、山梨醇69 g/L、甘露醇91 g/L。
上述培養基均于121℃滅菌20 min。
1.2 儀器與設備
SW-CJ-2FD型雙人單面凈化工作臺:蘇州凈化設備有限公司;WFJ7200型可見分光光度計:尤尼柯(上海)儀器有限公司;4380C型電轉儀、5804R低溫冷凍離心機:德國Eppendorf公司;GNP-9080型隔水式恒溫培養箱:上海精宏實驗設備有限公司;Veriti96孔熱循環梯度聚合酶鏈式反應(polymerasechain reaction,PCR)儀:美國應用生物系統公司;MD2000核酸超微量分光光度計:美國BioFure公司;ZQYZ-CS型恒溫振蕩培養箱:上海知楚儀器有限公司;DYY-12電泳儀:北京市六一儀器廠;UVIEssential V6凝膠成像儀:英國Uvitec公司。
1.3 實驗方法
1.3.1 引物設計
采用Oligo軟件設計引物,引物信息見表1。

表1 本實驗所用引物Table 1 Primers used in this experiment
1.3.2 同源重組片段spo0A-Cmr的制備
同源重組片段spo0A-Cmr的制備流程見圖1。

圖1 重組菌B.clausii QL-1△spo0A構建流程圖Fig.1 Flowchart of recombinant strain B.clausii QL-1△spo0A construction
spo0A基因的調取:挑取B.clausii QL-1單菌落轉接于50 mL LB液體培養基,37℃,200 r/min培養12 h,使用Ezup柱式基因組DNA抽提試劑盒對B.clausii QL-1菌體基因組進行提取。1μL基因組模板、1μL spo0A F和1μL spo0A R引物、12.5μL 2×HiFi-PCRmaster、9.5μLddH2O組成的25μL反應體系進行PCR擴增,獲得堿基長度為418 bp的spo0A基因片段,PCR擴增條件:95℃預變性5 min;95℃變性30 s,56℃退火30s,72℃延伸45s,30個循環;72℃再延伸10min,4℃保存。
片段Cmr基因的調取:挑取大腸桿菌JM109單菌落轉接于50 mL LB液體培養基,37℃,200 r/min培養12 h,使用高純度質粒小量提取試劑盒提取pHT01質粒。1μLpHT01質粒模版、1μL CmrF和1μL CmrR引物、12.5μL 2×HiFi-PCR master、9.5μL ddH2O組成的25μL反應體系進行PCR擴增,獲得長度為1 264 bp的Cmr抗性基因片段,PCR擴增條件:95℃預變性5 min;95℃變性30 s,58℃退火30 s,72℃延伸2 min35s,30個循環;72℃再延伸10min,4℃保存。
同源重組片段spo0A-Cmr的制備:使用SanPrep柱式DNA膠回收試劑盒回收所得的spo0A基因片段和Cmr抗性基因片段,首先進行第一步重疊PCR,2μL spo0A基因片段和2μL Cmr抗性基因片段互為模板及引物、12.5μL 2×HiFi-PCR master及3.5μL ddH2O組成的25μL反應體系進行重疊延伸PCR擴增,PCR擴增條件:95℃預變性5 min;95℃變性30 s,58℃退火30 s,72 ℃延伸2 min 35 s,8個循環;向第一步反應液中加入1μL spo0A F和1μL CmrR引物、12.5μL 2×HiFi-PCR master及10.5μL ddH2O進行第二步大量PCR擴增,PCR擴增條件:95℃預變性5 min;95℃變性30 s,58℃退火30 s,72℃延伸3 min 30 s,30個循環;72℃再延伸10 min,4℃保存。經過上述兩步重疊延伸PCR擴增得到1648bp的同源重組片段spo0A-Cmr。使用SanPrep柱式DNA膠回收試劑盒回收所得的spo0A-Cmr片段,-20℃保存備用。
1.3.3 同源重組片段spo0A-Cmr回收產物的酶切及濃縮
采用限制性核酸內切酶Bam HⅠ對所得的spo0A-Cmr片段進行酶切,30μL酶切體系:25μL PCR產物、1.5μL Bam HⅠ、3.5μL 10×K Buffer,37℃靜置1.5 h。向酶切產物中加入醋酸鈉(1/10體積,3mol/L)和無水乙醇(25倍體積),混合后-20℃處理20 min,12 000 r/min離心8 min得DNA沉淀;加入體積分數為70%乙醇重懸DNA沉淀,12 000 r/mim離心8 min,除去乙醇,37℃風干,加入ddH2O重懸,制得300~500 ng/μL的spo0A-Cmr濃縮DNA溶液。
1.3.4 克勞氏芽孢桿菌B.clausii QL-1感受態的制備
電轉緩沖液配制:山梨醇91 g/L、甘露醇91 g/L、甘油100 g/L。
將B.Clausii QL-1單菌落接種于15 mL的LB液體培養基中,37℃、200 r/min振蕩培養12 h;將上述菌液以2%接種量轉接到50 mL LB培養基中,37℃、200 r/min振蕩培養至OD600nm值為1.0;4 ℃、8 000 r/min離心8 min收集菌體;電轉緩沖液洗滌3~4次后重懸、分裝,即為制備好的感受態。感受態制備過程中均保證4℃條件下操作。
1.3.5 重組菌B.clausii QL-1△spo0A的構建
25 mg/mL氯霉素的制備:準確稱取氯霉素溶解于無水乙醇,溶解后過0.22μm濾膜過濾除菌,分裝后于-20℃中保存。
感受態與spo0A-Cmr濃縮片段以10∶1(V/V)比例混合,1500 V、5 ms條件下進行電轉化,立即加入1 mL的菌體復蘇培養基,37℃、180 r/min復蘇4~5 h后涂布于氯霉素終質量濃度為25μg/mL的LB固體培養基中培養18~24 h,篩選具有氯霉素抗性的菌株。
1.3.6 陽性重組菌株的鑒定
以提取到的陽性菌株DNA為模板,spo0A F和CmrR為引物PCR擴增,擴增產物進行1%瓊脂糖凝膠電泳分析,最終獲得陽性重組菌株B.clausii QL-1△spo0A。B.clausii QL-1△spo0A重組菌于LB液體培養基中,在37℃、200r/min振蕩培養條件下傳代15次,進行菌落PCR檢測。
1.3.7 出發菌株及重組菌株芽孢形成檢測
將菌株B.clausii QL-1與B.clausii QL-1△spo0A分別以2%接種量接種于LB液體培養基,37℃、200 r/min振蕩培養28 h,將菌液于80℃水浴處理10 min。將80℃水浴處理過的出發菌株菌液稀釋10-6倍、重組菌株菌液不稀釋,同樣分別做三個平行,取100μL涂布于LB固體平板上,將未處理的出發菌株及重組菌株的菌液分別稀釋10-6倍,做3個平行,取100μL涂布于LB固體平板上,上述平板放置于37℃培養12~18 h,用平板計數法觀察并清點100μL處理菌液及未處理菌液的芽孢數及菌體數(CFU/mL)。芽孢生成率計算公式如下:

1.3.8 B.clausii QL-1△spo0A重組菌生長曲線及生物量的測定
生長曲線的繪制:挑取菌株B.clausii QL-1與B.clausii QL-1△spo0A單菌落于15mLLB液體培養基,37℃、200r/min振蕩培養12 h,將菌液轉接至100 mL LB液體培養基至初始OD600nm值為0.1左右,繼續培養,每隔4 h取樣4 mL,使用紫外分光光度計在波長600 nm處測定吸光度值。
生物量的測定:每隔12h取樣10 mL,離心得菌體沉淀,于70℃烘箱烘干至恒質量后測其生物量。
1.3.9 B.clausii QL-1△spo0A重組菌淀粉酶酶活的測定
分別將出發菌株B.clausii QL-1和重組菌株B.clausii QL-1△spo0A單菌落點涂于可溶性淀粉平板中央,于37℃條件下靜置培養36 h,觀察并比較透明圈大小。分別以2%接種量接種在LB液體培養基中,37℃、200 r/min振蕩培養,每隔6 h取樣5 mL,4℃、12 000 r/min離心8 min取上清,按照國標GB/T 24401—2009《α-淀粉酶制劑》中所示方法測定淀粉酶酶活,并按干質量進行換算為U/g。
淀粉酶酶活定義:于60℃、pH值為6.0條件下,1 h液化1 g可溶性淀粉的酶量,即為一個酶活力單位(U/g)。
2 結果與分析
2.1 spo0A基因缺失菌株B.clausii QL-1△spo0A的構建
以B.clausii QL-1出發菌基因組為模板,spo0A F和spo0A R為引物進行PCR擴增,1%瓊脂糖凝膠電泳檢測,結果見圖2a。由圖2a可知,擴增產物大小為500 bp左右,與理論長度418 bp相符,說明成功獲得spo0A片段;以質粒pHT01為模板,CmrF和CmrR為引物PCR擴增,1%瓊脂糖凝膠電泳檢測,結果見圖2b。由圖2b可知,擴增產物大小為1000bp左右,與理論長度1 264 bp相符,說明成功獲得抗性基因Cmr片段;以膠回收所得的spo0A片段和Cmr片段為模板,spo0A F和CmrR為引物進行重疊延伸PCR擴增,1%瓊脂糖凝膠電泳檢測,結果見圖2c。由圖2c可知,擴增產物大小為1 500 bp左右,與理論長度1 648 bp相符,說明成功獲得同源重組片段spo0A-Cmr。將spo0A-Cmr片段經Bam HⅠ酶切后進行濃縮,測其質量濃度為450ng/μL。將10μL回收片段與100μL的B.clausii感受態混合進行電轉化,涂布于氯霉素抗性LB平板上,挑取陽性轉化子,以spo0A F和CmrR為引物進行菌落PCR驗證,1%瓊脂糖凝膠電泳檢測結果見圖2d。由圖2d可知,出現特異性目的條帶,表明成功獲得B.clausii QL-1△spo0A重組菌。
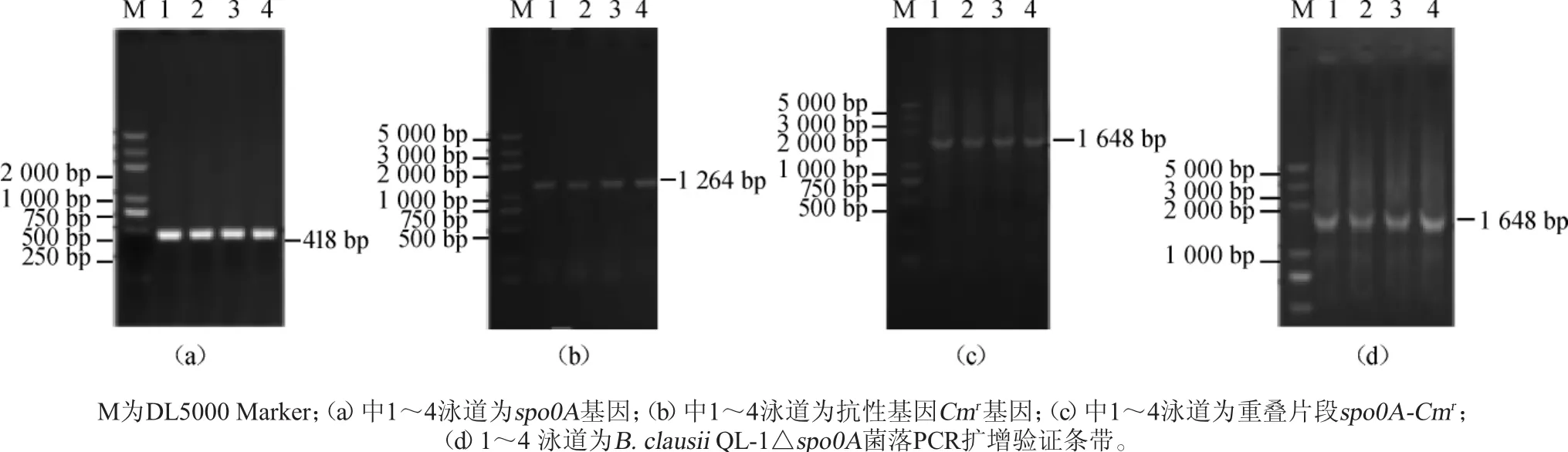
圖2 spo0A-Cmr片段PCR擴增產物及菌落PCR擴增產物凝膠電泳圖Fig.2 Electrophoresis of PCR amplified products of spo0A-Cmr and colony
通過連續傳代15次,以CmrF和CmrR為引物進行菌落PCR擴增,1%瓊脂糖凝膠電泳檢測,結果見圖3。由圖3可知,得到擴增產物大小為1 000 bp左右,與理論長度1 264 bp相符,說明B.clausii QL-1△spo0A成功導入Cmr抗性基因,以spo0A F和CmrR為引物得到的擴增產物大小同樣于1648bp出現特異性目的條帶,表明重組菌株遺傳穩定性較好。

圖3 B.clausii QL-1△spo0A重組菌株PCR擴增產物凝膠電泳圖Fig.3 Electrophoresis of PCR amplified products of recombinant strain B.clausii QL-1△spo0A
2.2 spo0A基因缺失克勞氏芽孢桿菌對芽孢萌發的影響
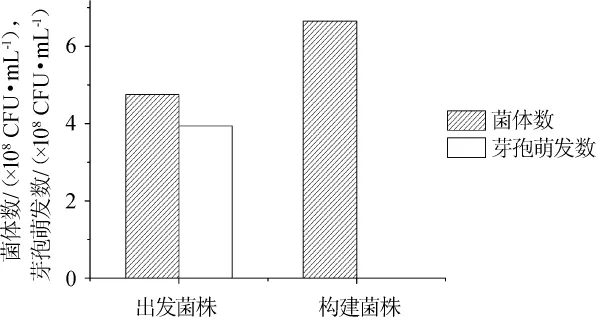
圖4 spo0A基因缺失對克勞氏芽孢桿菌芽孢萌發的影響Fig.4 Effect of spo0A gene deletion on spore germination of B.clausii
通過平板計數法對出發菌株及重組菌株芽孢生成情況進行計算及比較,結果見圖4。由圖4可知,菌株B.clausii QL-1芽孢萌發數為3.93×108CFU/mL,菌體數為4.67×108CFU/mL,計算得到出發菌株B.clausii QL-1芽孢生成率約為84.15%,通過平板計數法及顯微鏡觀察,B.clausii QL-1△spo0A均不生成芽孢。結果表明,spo0A基因是克勞氏芽孢桿菌芽孢萌發過程中的關鍵基因。
2.3 spo0A基因缺失克勞氏芽孢桿菌對菌株生長的影響
對出發菌株B.clausii QL-1及重組菌株B.clausii QL-1△spo0A進行了生長規律分析及生物量測定,結果見圖5。

圖5 spo0A基因缺失對菌株在LB培養基中生長周期(a)及生物量(b)的影響Fig.5 Effect of spo0A gene deletion on the growth cycle(a)and biomass(b) of strain in LB medium
由圖5a可知,B.clausii QL-1△spo0A菌株與出發菌株的生長周期基本保持一致,4~18 h為對數生長期,18~28 h為穩定期,28 h后進入衰亡期。由圖5b可知,在任何時間段,重組菌株B.clausii QL-1△spo0A生物量均高于出發菌株B.clausii QL-1,且均在20 h出現最大生物量,分別為3.10 mg/mL、2.50 mg/mL,生物量提高了24%。
2.4 spo0A基因缺失克勞氏芽孢桿菌對淀粉酶分泌的影響
可溶性淀粉平板初篩,重組菌株B.clausii QL-1△spo0A較出發菌株B.clausii QL-1透明圈增大,重組菌株透明圈與菌落直徑比為2.75,出發菌株透明圈與菌落直徑比為1.31,說明構建菌株較出發菌株水解淀粉能力更強。進一步于LB液體培養基發酵培養并測定淀粉酶酶活,結果見圖6。由圖6可知,重組菌株B.clausii QL-1△spo0A產淀粉能力高于出發菌株B.clausii QL-1菌株,且發酵至72 h時,淀粉酶酶活均達到最高,分別為1.58×105U/g、0.86×105U/g,較出發菌株B.clausii QL-1,重組菌株B.clausii QL-1△spo0A淀粉酶酶活提高83.72%。

圖6 spo0A基因缺失對淀粉酶酶活的影響Fig.6 Effect of spo0A gene deletion on amylase activity
3 結論
本實驗室通過同源單交換技術成功實現了克勞氏芽孢桿菌spo0A基因的敲除,構建了spo0A基因缺失型菌株B.clausii QL-1△spo0A,并對其發酵性能進行初步研究,發現spo0A基因缺失型菌株不生成芽孢,且較出發菌株B.clausii QL-1生物量提高24%,表明spo0A基因不僅是克勞氏芽孢桿菌芽孢形成關鍵基因,而且也是菌株生長的重要基因,為構建芽孢缺失型克勞氏芽胞桿菌工業生產菌株奠定了基礎。通過對B.clausii QL-1△spo0A菌株的進一步研究發現,重組菌株B.clausii QL-1△spo0A較出發菌株B.clausii QL-1淀粉酶酶活提高83.72%,表明spo0A基因也是克勞氏芽孢桿菌淀粉酶生成的重要相關基因,該發現對高產淀粉酶芽孢桿菌類微生物的構建具有指導意義。
