蛋白體外結合實驗聯合質譜分析鑒定Num1的互作蛋白
2018-12-29 00:52:02唐仙英
中南民族大學學報(自然科學版) 2018年4期
唐仙英,肖 瑤,海 力
(中南民族大學 生命科學學院,武陵山區特色資源植物種質保護與利用湖北省重點實驗室,武漢430074)
為確保細胞的命運決定因子準確地分配到每個子細胞,所有真核細胞均須正確定位有絲分裂紡錘體[1].細胞質動力蛋白Dynein在各類細胞的紡錘體定位過程中起關鍵作用[2-4].在芽殖酵母的不對稱分裂過程中,為了能沿胞質微管產生拉力移動紡錘體,Dynein在與微管相連的同時,需被錨定在細胞膜上.Dynein在細胞膜上的錨定依賴于Num1(Nuclear migration 1,核遷移1)[5-7],Num1是迄今為止所發現的參與紡錘體定位的唯一位于細胞膜上的因子.Num1蛋白大約313 kDa,由多個結構域組成.其中,位于其C末端的PH結構域通過與細胞膜上的磷酸肌醇PI(4,5)P2相互作用介導Num1與細胞膜結合[8,9];而另一個位于其N末端含有兩段預測的Coiled-coil序列的結構域則通過自我相互作用介導Num1在細胞膜上形成由14個分子組成的補丁狀復合物,因此被命名為Patch Assembly(PA)結構域[10].此外,PA結構域也介導Num1與Dynein的相互作用[10].
定位于細胞膜上的Num1復合物不具有運動性[8, 11],除了在紡錘體定位中的作用,它還通過與線粒體膜上特定種類的脂類物質結合而介導線粒體與細胞膜的連接,并由此調節線粒體的分裂[12,13].Lackner 等[10, 12]發現線粒體與細胞膜的連接是由Num1的PA結構域尤其是其中預測的Coiled-coil序列所介導,但Num1在紡錘體定位與線粒體中的活性如何調節目前尚不清楚.本研究利用在BL21大腸桿菌細胞中表達純化的PA結構域,通過Pull-down實驗分離酵母細胞中與PA結構域相互作用的蛋白質,并進一步通過質譜分析完成鑒定,旨在通過鑒定Num1的互作蛋白,為揭示Num1的作用途徑及機制奠定基礎.
1 材料與方法
1.1 材料和儀器
DH5α和BL21大腸桿菌細胞(北京鼎國昌盛);蛋白酶抑制劑混合物(Protease Inhibitor Cocktail, Roche),S蛋白瓊脂糖(S protein agarose slurry)、Bugbuster蛋白提取反應液(Novagen);氨芐青霉素、氯霉素、異丙基硫代半乳糖苷IPTG、苯甲基磺酰氟PMSF(BioSharp).
高速冷凍離心機(CR22G型, HITACHI);微量冷凍離心機(FC5515R型, OHAUS);高壓細胞破碎儀(BT40/TS2/AA型, Constant System Cell Disruptor);凝膠成像分析儀(Universal Hood II型, BIO-RAD).
1.2 質粒與酵母菌株
編碼Num1PA(1-303)-PCN-S-TEV-Z的質粒pXT65(PCN: PreScission蛋白酶酶切位點,S: S-tag, TEV: TEV蛋白酶酶切位點,Z: IgG binding motif)用于表達PA結構域與S-tag的融合蛋白,其構建方法如下:編碼Num1 1-303 aa且在5′端含有一個NcoI限制性內切酶位點,3′端含有一個NotI限制性內切酶位點的DNA片段分別用正向引物5′-CCAGCCATGGCCTCCCACAACAACAGGCATAAAAAG-3′和反向引物5′-CCAGGCGGCCGCCAGATGTTACTGTAGTATCG-3′從酵母基因組DNA通過PCR擴增,擴增的片段經NcoI和NotI酶切后與同樣經NcoI和NotI酶切的載體pBSG01[10]連接并經測序確認,產生pXT65.pXT66(PCN-S-TEV-Z)為空載體對照.酵母菌株YWL555[10].
基因型為MATαnum1Δ∷HIS3ura3-52lys2-801leu2-Δ1his3-Δ200trp1-Δ63.
1.3 大腸桿菌細胞培養
質粒制備或轉化用DH5α細胞,重組蛋白表達和制備用BL21細胞[14].DH5α細胞用含氨芐青霉素的LB培養基于37 ℃培養.用于制備重組蛋白的BL21細胞按下述方式培養:保存于-80 ℃的BL21大腸桿菌細胞在含氨芐青霉素的LB固體培養基上37 ℃過夜培養.次日從LB平板上挑取細胞接種到含氨芐青霉素和氯霉素的LB液體培養基,37 ℃過夜培養.第3 d將液體培養基中的細胞稀釋至OD600=0.1,25 ℃培養3 h,加入異丙基硫代半乳糖苷(IPTG)至終濃度為0.5 mmol/L,20 ℃培養16 h誘導融合蛋白表達,離心收集細胞.
1.4 蛋白可溶性檢測
按1.2培養的BL21細胞經離心收集后,按每0.01 g細胞加入50 μL Bugbuster蛋白提取反應液,混勻,室溫下輕搖15 min,4 ℃ 16000 g 離心20 min;分別取上清和沉淀加入蛋白上樣緩沖液,沸水煮5 min.將上清和沉淀制備的蛋白樣品分別經變性聚丙烯酰胺凝膠電泳(SDS-PAGE)分離后用考馬斯亮蘭染色,比較目的蛋白在上清和沉淀中的分配比例.
1.5 Pull-down實驗
1.5.1 重組蛋白的制備
攜帶pXT65和pXT66的BL21大腸桿菌按1.3各收集5 g細胞.按照每克細胞1 mL的比例加入Bind/Wash buffer [150 mmol/L NaCl, 20 mmol/L Tris-Cl (pH 7.5), 0.05% TritonX-100, 1 mmol/L EDTA, 1 mmol/L DTT, Protease Inhibitor Cocktail],用高壓細胞破碎儀(壓力207 MPa, 4 ℃)破碎細胞.破碎的細胞4 ℃下12000 g 離心10 min,收集上清,此為大腸桿菌細胞裂解液.取100 μL S protein agrose懸液,1000 g離心1 min,除去上清,用Bind/Wash buffer洗3次,每次1 mL.加入大腸桿菌細胞裂解液,4 ℃旋轉混合45 min,離心,除去上清.再用Bind/Wash buffer洗珠子3次,每次5 mL,洗過的珠子置于冰上.
1.5.2 酵母細胞裂解液的制備
離心收集YPD液體培養基中旺盛生長的對數期YWL555細胞,每個pull-down實驗需要5 g濕細胞. 按1 mL/g的比例加入酵母裂解緩沖液[30 mmol/L HEPES (pH 7.4), 50 mmol/L乙酸鉀, 2 mmol/L醋酸鎂, 0.2 mmol/L EGTA, 0.05% Triton X-100, 1 mmol/L DTT, Protease Inhibitor Cocktail (Roche)],用高壓細胞破碎儀破碎細胞.破碎的細胞液10000 g離心5 min,上清即為酵母細胞裂解液.
1.5.3 Pull-down反應
將1.5.2中制備的酵母細胞裂解液加入到1.5.1中已結合了重組蛋白的S蛋白瓊脂糖中,4 ℃旋轉2 h,1000 g離心1 min,移去上清.用Wash buffer [10 mmol/L Tris-Cl (pH 8.0), 150 mmol/L KCl, 10% Glycerol, 0.05% Triton X-100, 1 mmol/L DTT, Protease Inhibitor Cocktail (Roche)] 洗3次,每次8 mL,除去Wash buffer,加入100 μL 蛋白上樣緩沖液,沸水煮5 min,收集上清保存于-20℃.
1.6 SDS-聚丙烯酰胺凝膠電泳及蛋白鑒定
所有的蛋白質樣品均用10%變性聚丙烯酰胺凝膠電泳進行檢測[15].其中Pull-down沉淀下來的酵母蛋白樣品10% SDS-聚丙烯酰胺凝膠電泳分離后,再用考馬斯亮藍染色液染色,切下被PA結構域融合蛋白沉淀下來的特異性蛋白條帶進行鳥槍法(shotgun)LC-MS質譜分析.
2 結果與分析
2.1 PA結構域重組蛋白的表達
PA結構域是預測的Coiled-coil結構,高度不可溶.比較了不同培養條件下Num1PA(1-303)-PCN-S-TEV-Z融合蛋白在BL21大腸桿菌中的表達(見圖1),最后確定適宜的誘導條件為IPTG濃度0.5 mmol/L,20 ℃過夜培養,此時Num1PA(1-303)-PCN-S-TEV-Z存在于細胞裂解液上清的比例相對較高,圖1中虛線框對應預期蛋白帶,箭頭指示為該蛋白在20 ℃條件下表達(圖1b)較37 ℃下表達(圖1a)時存在于裂解液上清中的比例高.
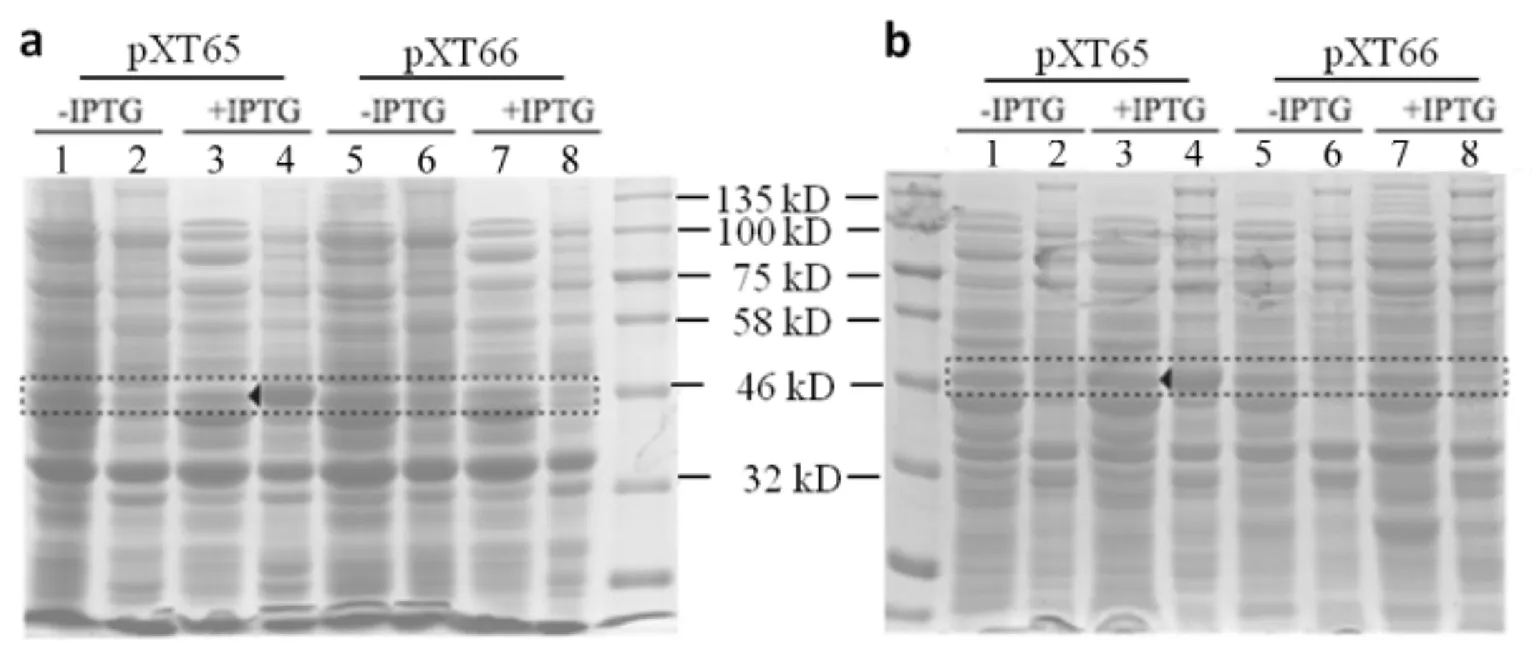
1,3,5,7)上清;2,4,6,8) 沉淀a) 37 ℃, IPTG 0.5 mmol/L; b) 20 ℃, IPTG 0.5 mmol/L圖1 不同條件下蛋白誘導表達結果Fig.1 Protein expression induced under different conditions
2.2 PA結構域重組蛋白的純化
攜帶pXT65[Num1PA(1-303)-PCN-S-TEV-Z]或pXT66(PCN-S-TEV-Z)的BL21大腸桿菌細胞在含0.5 mmol/L IPTG濃度的培養基中20 ℃過夜培養誘導蛋白表達.由于誘導表達的Num1PA(1-303)-PCN-S-TEV-Z蛋白含有一個S-tag,因此可在裂解大腸桿菌細胞后,使其與S蛋白瓊脂糖珠子結合而得到純化,結果見圖2.如圖2所示:S蛋白瓊脂糖能從表達pXT65的大腸桿菌細胞裂解液中沉淀下來Num1PA(1-303)-PCN-S-TEV-Z蛋白(箭頭所示),而未從表達空載體pXT66的大腸桿菌細胞裂解液中沉淀下來蛋白.
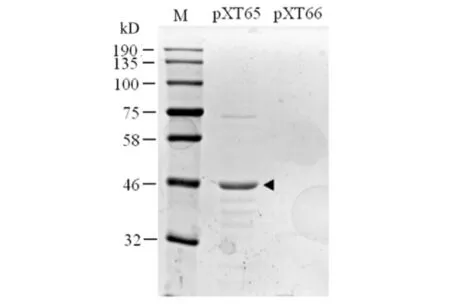
圖2 Num1PA(1-303)-PCN-S-TEV-Z蛋白的純化Fig.2 Purification of Num1PA(1-303)-PCN-S-TEV-Z protein
2.3 PA結構域重組蛋白與酵母蛋白的離體結合
為分離與PA結構域相互作用的酵母蛋白,用結合在S蛋白瓊脂糖珠子上的Num1PA(1-303)-PCN-S-TEV-Z蛋白與酵母細胞裂解液進行了離體結合實驗,即Pull-down實驗.由于PA結構域具有自我相互結合的特性[10],為防止PA結構域與酵母內源的Num1蛋白相結合,酵母細胞裂解液從已敲除了野生型NUM1基因的酵母菌株(num1Δ,即YWL555)中制備.洗去非特異性結合的蛋白質,即獲得了結合在S蛋白瓊脂糖珠子上的Num1PA(1-303)-PCN-S-TEV-Z及與其結合的其他蛋白質的復合物.此蛋白復合物樣品經SDS-PAGE分離后用考馬斯亮藍染色,結果見圖3.如圖3所示:上箭頭為Num1PA(1-303)-PCN-S-TEV-Z蛋白,下箭頭是被Num1PA(1-303)-PCN-S-TEV-Z特異性沉淀下來的蛋白復合物(即pXT66的Pull-down樣品中缺乏的蛋白),將其切割下來進行質譜分析,用于鑒定與PA結構域互作的蛋白.

圖3 Num1PA(1-303)-PCN-S-TEV-Z的Pull-down蛋白檢測
Fig.3 Detection of proteins pulled down by Num1PA(1-303)-PCN-S-TEV-Z
2.4 互作蛋白的鑒定
通過對Pull-down沉淀下來的蛋白復合物進行鳥槍法(shotgun)LC-MS質譜分析,鑒定出一系列新的與Num1相互作用的蛋白,按照參與的細胞功能分組如表1.
3 結語
位于Num1 N末端的PA結構域(1-303 aa)在Num1的功能中起核心作用,缺失了PA結構域的Num1將同時失去其在紡錘體定位和線粒體中的功能[10,12,13].為分離Num1的互作蛋白,在大腸桿菌細胞中表達了PA結構域和純化標簽的融合蛋白Num1PA(1-303)-PCN-S-TEV-Z,并用S protein agarose對其進行了純化.結合在S protein agarose珠子上的重組蛋白用于從不表達內源Num1蛋白的酵母細胞裂解液中沉淀PA結構域的互作蛋白.通過對沉淀下來的蛋白進行鳥槍法LC-MS質譜分析,鑒定了一系列新的Num1的互作蛋白,包括大量的核糖體蛋白和參與蛋白折疊轉運的蛋白,參與基因轉錄和翻譯的核酸酶和蛋白酶,及其他功能各異的蛋白質.這些結果為進一步揭示Num1在紡錘體定位及線粒體中的作用機制奠定了基礎,也為此過程的調控機制提供了更多思路.

表1 Num1的互作蛋白Tab.1 Proteins interacting with Num1
