UV法或HPLC法結合金屬離子沉淀法測定米索硝唑pH敏感脂質體中主成分含量的比較Δ
2019-01-02 03:19:44魏巍何美王琛李睿李必波羅治彬重慶市人民醫院腫瘤血液科重慶000西南醫科大學附屬醫院腫瘤科四川瀘州66000重慶市腫瘤研究所腫瘤防治辦公室重慶0000重慶藥友制藥有限責任公司重慶02
中國藥房 2018年24期
魏巍,何美,王琛,李睿,李必波#,羅治彬,2(.重慶市人民醫院腫瘤血液科,重慶 000;2.西南醫科大學附屬醫院腫瘤科,四川 瀘州 66000;.重慶市腫瘤研究所腫瘤防治辦公室,重慶 0000;.重慶藥友制藥有限責任公司,重慶 02)
米索硝唑是一種具有較高放射增敏比的乏氧細胞放射增敏劑,其放射增敏比為1.5[1],最高可增至1.8~2.0[2],常用于頭頸部鱗狀細胞癌的放療增敏。但因其水溶性低、油水分配系數高導致其不良反應發生率高,主要為外周神經系統毒性(發生率為15%~28%),故而限制了其臨床應用[3-4]。為此,本課題組將米索硝唑包裹于pH敏感脂質體中制成米索硝唑pH敏感脂質體,以此來克服藥物的外周神經毒性,并使其具有腫瘤靶向性[5-8]。
藥物含量測定是制劑質量控制中非常重要的項目。既往米索硝唑相關檢測均采用高效液相色譜法(HPLC)測定原藥或其代謝產物[9-10],該法準確、可靠,但對儀器的要求較高,且測定過程煩瑣、耗時、耗材。相比之下,紫外分光光度法(UV)操作簡單、費用更低,且在一定質量濃度范圍內檢測較為準確。然而,本課題組前期研究發現,米索硝唑pH敏感脂質體中的輔料磷脂酰乙醇胺對UV法測定藥物含量的干擾大。如能通過簡單的物理或化學預處理法,將脂質體中的磷脂酰乙醇胺除去,則可為UV法的應用奠定基礎。為此,本研究擬采用金屬離子沉淀法(以ZnCl2為金屬離子沉淀劑)預處理除去米索硝唑pH敏感脂質體中的磷脂酰乙醇胺,進而采用UV法測定其中主成分的含量,并將結果與HPLC法比較,為實現快速、簡便、準確、低廉地測定該制劑中主成分的含量提供理論依據。
1 材料
1.1 儀器
1100型HPLC儀,包括VWD紫外檢測器等(美國Agilent公司);ML204型電子分析天平(瑞士Mettler-Toledo公司);UV-2400PCS型紫外-可見分光光度計(上海舜宇恒平科學儀器有限公司);PHS-3C型pH計(上海三信儀表廠);RE-52AA型旋轉蒸發器(上海振捷實驗設備有限公司);SHZ-D(Ⅲ)型循環水式多用真空泵(河南予華儀器制造有限公司);XW-80A型旋渦混合器(上海醫科大學儀器廠);SK1200H型超聲波清洗器(上海科導超聲儀器有限公司);TGL-16G型臺式離心機(上海安亭科學儀器廠);ZRS-8G型智能溶出儀(天津大學無線電廠)。
1.2 藥品與試劑
米索硝唑原料藥(青島捷世康生物科技有限公司,批號:20170219,純度:≥98.5%);米索硝唑對照品(美國Cayman公司,批號:15606-201610,純度:99.5%);磷脂酰乙醇胺、膽固醇、亞油酸、維生素E、羧甲基殼聚糖(羧化度:80%)、ZnCl2均為藥用級;甲醇為色譜純,其余試劑均為分析純,水為雙蒸水。
2 方法與結果
2.1 米索硝唑pH敏感脂質體的制備
采用薄膜分散法制備米索硝唑pH敏感脂質體。按處方輔料配比分別精密稱取磷脂酰乙醇胺、膽固醇、亞油酸、維生素E(比例為3∶1∶1∶0.1,m/m/m/m)各適量,置于同一100 mL圓底燒瓶中,加入10 mL氯仿使溶解,將燒瓶連接旋轉蒸發器,在37℃、100 r/min、減壓條件下揮干氯仿,得貼于燒瓶內壁一層均勻的薄膜;加入含米索硝唑的磷酸鹽緩沖液(PBS,pH 7.4)(藥液比為1∶9,m/m)使溶解,在50℃、100 r/min、避光條件下水化60 min,得微黃色脂質體溶液;將上述脂質體溶液超聲(功率:180 W,頻率:40 kHz)處理10 min,經0.22 μm微孔濾膜濾過,取續濾液,加入0.5 mL 0.3%羧甲基殼聚糖溶液,在50℃、100 r/min、避光條件下繼續水合0.5 h,即得米索硝唑pH敏感脂質體。另不加入米索硝唑,同法制備空白脂質體。取上述空白脂質體5 mL,按處方比例加入適量米索硝唑溶液,物理混勻,即得空白脂質體+米索硝唑溶液。
2.2 溶液的制備
2.2.1 對照品溶液 精密稱取在五氧化二磷中干燥12 h后的米索硝唑對照品12.0 mg,置于25 mL量瓶中,加甲醇溶解并定容,搖勻,制成米索硝唑質量濃度為0.48 mg/mL的對照品溶液。
2.2.2 輔料溶液 按處方輔料配比分別精密稱取磷脂酰乙醇胺、膽固醇、亞油酸、維生素E各適量,先以氯仿溶解,隨后加入甲醇稀釋,制成磷脂酰乙醇胺、膽固醇、亞油酸、維生素E質量濃度分別為2.33、0.82、0.84、0.08 mg/mL的混合溶液;羧甲基殼聚糖以水溶解制成質量濃度為0.02 mg/mL的溶液。將上述2種溶液等體積混合,即得。
2.2.3 未經預處理的空白脂質體溶液 精密量取“2.1”項下空白脂質體100 μL,加入900 μL甲醇破乳,渦旋5 min混勻,室溫靜置15 min,12 000 r/min離心5 min,取上清液,即得。
2.2.4 經預處理的米索硝唑pH敏感脂質體溶液 精密量取“2.1”項下米索硝唑pH敏感脂質體100 μL,加入800 μL甲醇破乳,渦旋5 min混勻,加入50 mg/mL ZnCl2甲醇溶液100 μL,渦旋5 min混勻,室溫靜置15 min,12 000 r/min離心5 min,取上清液,即得。
2.2.5 經預處理的空白脂質體溶液 精密量取“2.1”項下空白脂質體 100 μL,加入800 μL甲醇破乳,渦旋5 min混勻,加入50 mg/mL ZnCl2甲醇溶液100 μL,渦旋5 min混勻,室溫靜置15 min,12 000 r/min離心5 min,取上清液,即得。
2.2.6 經預處理的空白脂質體+米索硝唑溶液 精密量取“2.1”項下空白脂質體+米索硝唑100 μL,加入800 μL甲醇破乳,渦旋5 min混勻,加入50 mg/mL ZnCl2甲醇溶液100 μL,渦旋5 min混勻,室溫靜置15 min,12 000 r/min離心5 min,取上清液,即得。
2.3 UV法測定脂質體中主成分的含量
2.3.1 專屬性試驗 (1)未經預處理樣品。取“2.2”項下對照品溶液、輔料溶液、未經預處理的空白脂質體溶液各適量,于200~400 nm波長范圍內掃描,光譜圖見圖1(圖中PE為磷脂酰乙醇胺,LA為亞油酸,VE為維生素E,CHO為膽固醇,CMC為羧甲基殼聚糖)。由圖1可知,米索硝唑在322 nm波長處有最大吸收;但輔料磷脂酰乙醇胺在322 nm波長處也有較大吸收,即對主成分測定有很大干擾,而輔料膽固醇、亞油酸、維生素E、羧甲基殼聚糖在322 nm波長處無明顯吸收,即對主成分測定無明顯干擾;未經預處理的空白脂質體在322 nm波長處亦有較明顯的紫外吸收,即此時無法消除磷脂酰乙醇胺對主成分測定的干擾。
(2)經預處理樣品。取“2.2”項下經預處理的空白脂質體溶液、空白脂質體+米索硝唑溶液、米索硝唑pH敏感脂質體溶液各適量,于200~400 nm波長范圍內掃描,光譜圖見圖2。由圖2A可知,經預處理完全沉淀了輔料磷脂酰乙醇胺,空白脂質體在322 nm波長處無明顯的紫外吸收;由圖2B、C可知,米索硝唑的最大吸收峰仍出現在322 nm波長處,因而此時輔料對主成分的測定已無干擾。
2.3.2 線性關系考察 分別精密量取“2.2.1”項下對照品溶液10、20、40、80、160、320 μL,置于5 mL量瓶中,加甲醇定容,搖勻,制成系列對照品溶液。以甲醇為空白,于322 nm波長處測定吸光度。以米索硝唑質量濃度(c,μg/mL)為橫坐標、吸光度(A)為縱坐標進行線性回歸,得米索硝唑回歸方程A=0.032 8c-0.008(r=0.999 6)。結果表明,米索硝唑檢測質量濃度線性范圍為0.96~30.72 μg/mL。
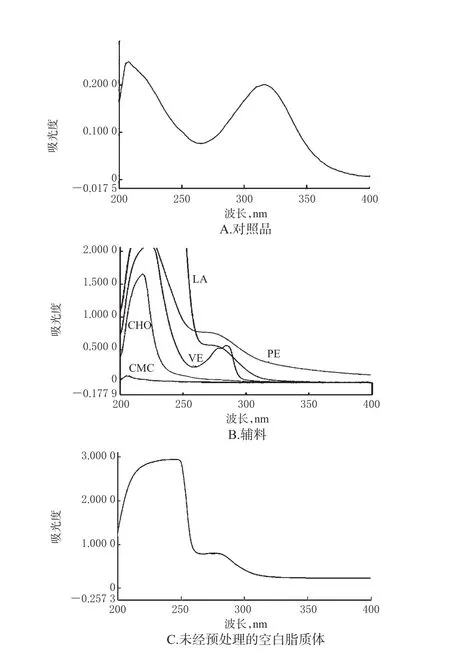
圖1 未經預處理樣品的紫外吸收光譜圖Fig 1 UV absorption spectra of untreated sample
2.3.3 精密度試驗 取“2.2.1”項下對照品溶液適量,分別加甲醇制成低、中、高質量濃度(3.84、7.68、15.36 μg/mL)對照品溶液,以甲醇為空白,于322 nm波長處測定吸光度,連續測定6次。結果,米索硝唑吸光度的RSD分別為0.93%、0.62%、0.48%(n=6),表明儀器精密度良好。
2.3.4 穩定性試驗 取“2.2.4”項下經預處理的米索硝唑pH敏感脂質體溶液適量,分別在室溫下放置0、2、4、6、12 h時,以甲醇為空白,于322 nm波長處測定吸光度。結果,米索硝唑吸光度的RSD為0.91%(n=5),表明經預處理的米索硝唑pH敏感脂質體溶液在室溫下放置12 h內基本穩定。
2.3.5 重復性試驗 取“2.1”項下米索硝唑pH敏感脂質體適量,共6份,按“2.2.4”項下方法制備經預處理的米索硝唑pH敏感脂質體溶液。以甲醇為空白,于322 nm波長處測定吸光度并計算主成分的含量。結果,米索硝唑含量平均值為99.20%,RSD為1.55%(n=6),表明本方法重復性良好。
2.3.6 回收率試驗 取“2.1”項下米索硝唑pH敏感脂質體適量,共9份,按“2.2.4”項下方法制成低、中、高質量濃度的米索硝唑pH敏感脂質體溶液。以甲醇為空白,于322 nm波長處測定吸光度并計算回收率,結果見表1。
2.3.7 米索硝唑pH敏感脂質體中主成分的含量測定 取“2.1”項下米索硝唑pH敏感脂質體適量,按“2.2.4”項下方法制備經預處理的米索硝唑pH敏感脂質體溶液。以甲醇為空白,于322 nm波長處測定吸光度并計算含量。平行測定3次。結果,3次測得的米索硝唑含量分別為99.86%、100.16%、100.32%(RSD=0.56)。
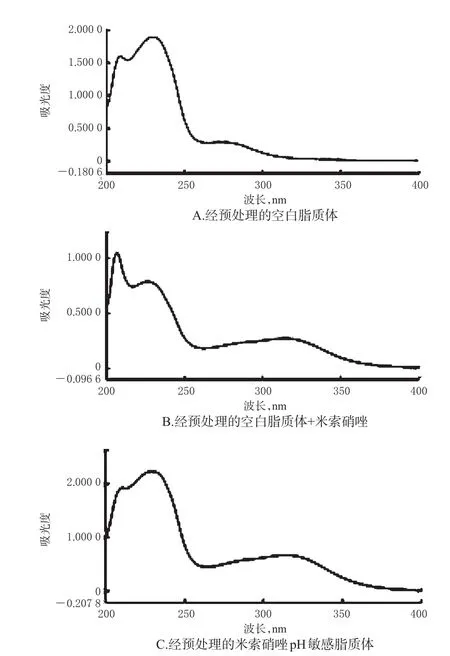
圖2 經預處理樣品的紫外吸收光譜圖Fig 2 UV absorption spectra of treated sample
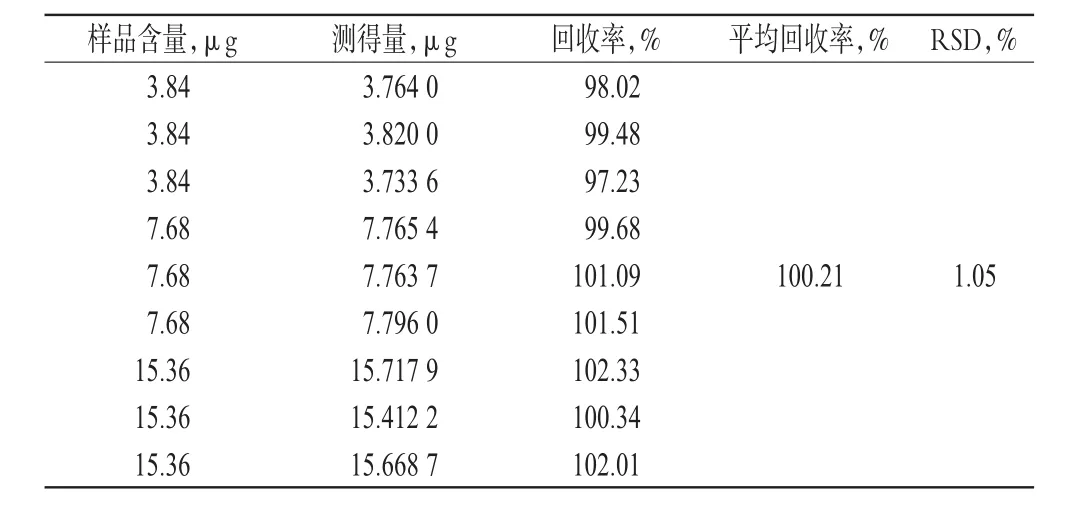
表1 UV法回收率試驗結果(n=9)Tab 1 Results of recovery tests for UV(n=9)
2.4 HPLC法測定脂質體中主成分的含量
2.4.1 色譜條件 色譜柱:Hypersil C18(250 mm×4.6 mm,5 μm);流動相:甲醇-水(20∶80,V/V);流速:1.0 mL/min;檢測波長:322 nm;柱溫:30 ℃;進樣量:20 μL。
2.4.2 專屬性試驗 取“2.2”項下對照品溶液、空白脂質體溶液、經預處理的米索硝唑pH敏感脂質體溶液和經預處理的空白脂質體+米索硝唑溶液各適量,按“2.4.1”項下色譜條件進樣測定,記錄色譜圖,詳見圖3。由圖3A可知,米索硝唑出峰時間為6.075 min;由圖3B可知,經預處理的空白脂質體已基本除去干擾主成分測定的輔料磷脂酰乙醇胺;由圖3C、D可知,經預處理的空白脂質體+米索硝唑和米索硝唑pH敏感脂質體藥物結構及極性未受影響,主成分出峰時間一致。
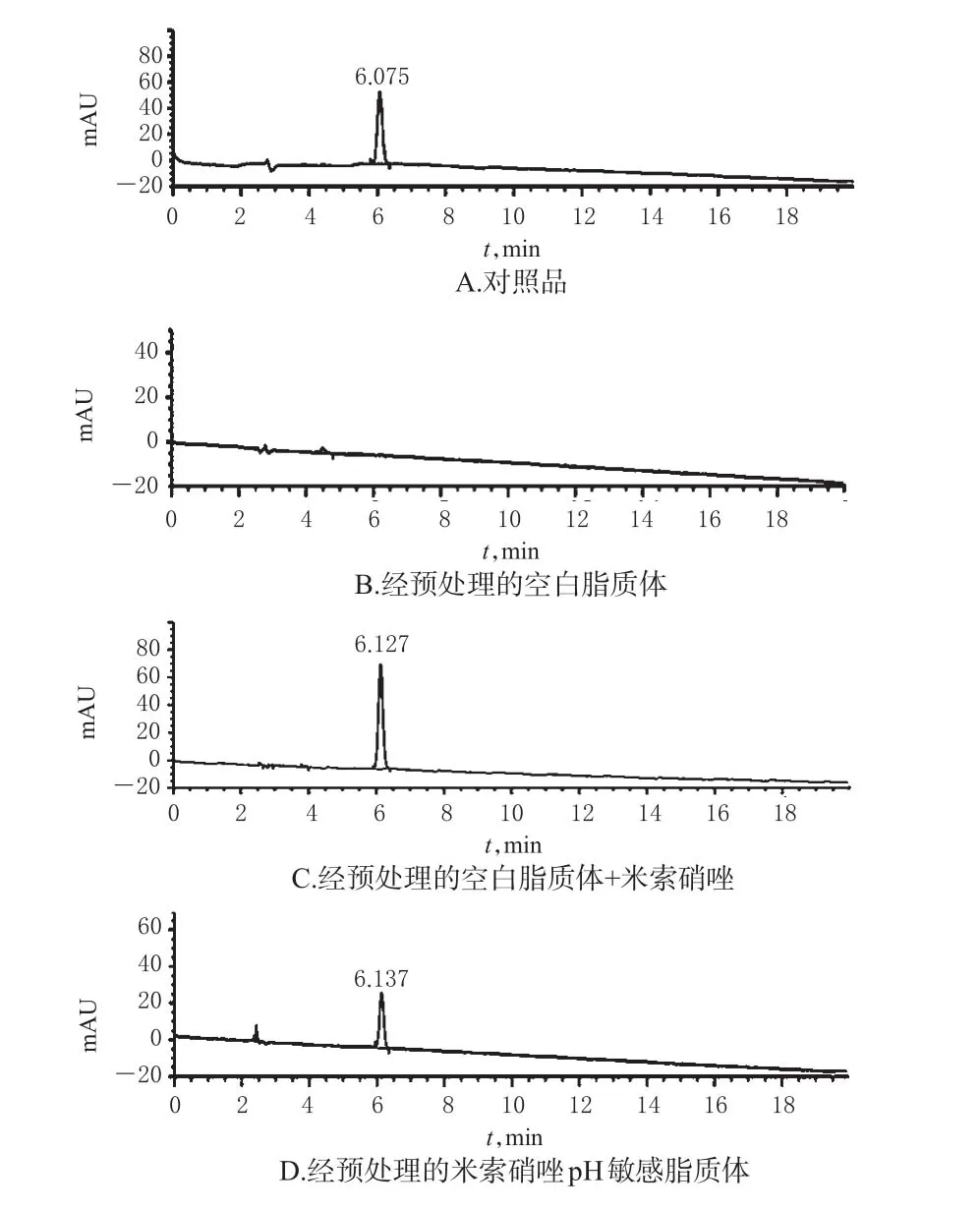
圖3 高效液相色譜圖Fig 3 HPLC chromatograms
2.4.3 線性關系考察 分別精密量取“2.2.1”項下對照品溶液5、10、20、30、40、50 μL,置于5 mL量瓶中,加甲醇定容,搖勻,制成系列對照品溶液。精密量取上述系列對照品溶液各20 μL,按“2.4.1”項下色譜條件進樣測定,記錄峰面積。以米索硝唑質量濃度(x,μg/mL)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得米索硝唑回歸方程y=56.82x-5.36(r=0.999 5)。結果表明,米索硝唑檢測質量濃度線性范圍為0.48~4.80 μg/mL。
2.4.4 定量限與檢測限考察 分別精密量取“2.2.1”項下對照品溶液適量,以甲醇倍比稀釋,按“2.4.1”項下色譜條件進樣測定,以信噪比10∶1、3∶1分別計算定量限、檢測限。結果,米索硝唑的定量限、檢測限分別為0.23、0.08 μg/mL。
2.4.5 精密度試驗 取“2.2.1”項下對照品溶液適量,按“2.4.1”項下色譜條件連續進樣測定6次,記錄峰面積。結果,米索硝唑峰面積的RSD為1.08%(n=6),表明儀器精密度良好。
2.4.6 穩定性試驗 取“2.2.4”項下經預處理的米索硝唑pH敏感脂質體溶液適量,分別在室溫下放置0、2、4、6、12 h時,按“2.4.1”項下色譜條件進樣測定,記錄峰面積。結果,米索硝唑峰面積的RSD為1.55%(n=5),表明經預處理的米索硝唑pH敏感脂質體溶液在室溫下放置12 h內基本穩定。
2.4.7 重復性試驗 取“2.1”項下米索硝唑pH敏感脂質體適量,共6份,按“2.2.4”項下方法制備經預處理的米索硝唑pH敏感脂質體溶液,再按“2.4.1”項下色譜條件進樣測定,記錄峰面積并計算主成分的含量。結果,米索硝唑含量平均值為99.60%,RSD為1.38%(n=6),表明本方法重復性良好。
2.4.8 回收率試驗 取“2.1”項下米索硝唑pH敏感脂質體適量,共9份,按“2.2.4”項下方法制成低、中、高質量濃度的米索硝唑pH敏感脂質體溶液,再按“2.4.1”項下色譜條件進樣測定,記錄峰面積并計算回收率,結果見表2。
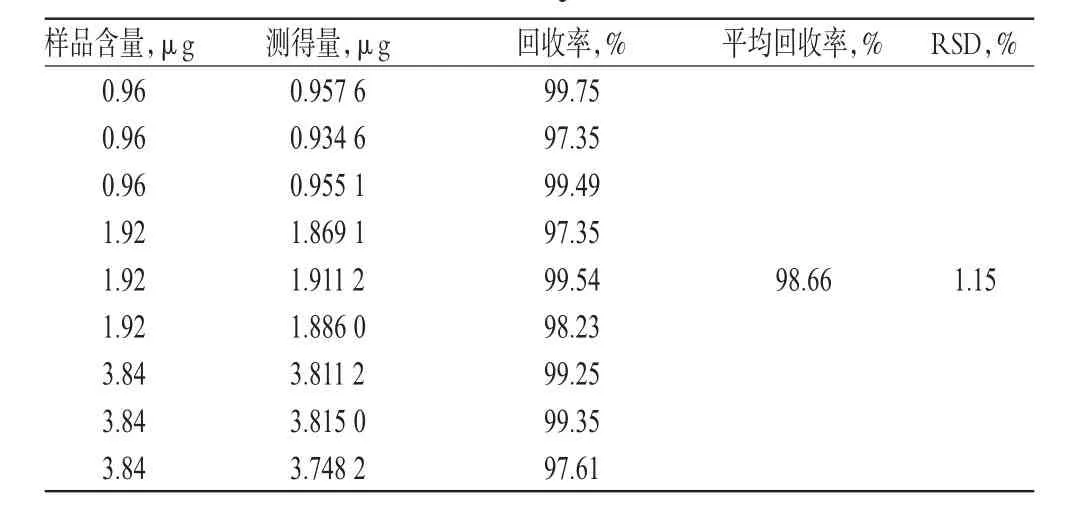
表2 HPLC法回收率試驗結果(n=9)Tab 2 Results of recovery tests for HPLC(n=9)
2.4.9 米索硝唑pH敏感脂質體中主成分的含量測定 取“2.1”項下米索硝唑pH敏感脂質體適量,按“2.2.4”項下方法制備經預處理的米索硝唑pH敏感脂質體溶液,再按“2.4.1”項下色譜條件進樣測定并計算主成分的含量。平行測定3次。結果,3次測得的米索硝唑含量分別為99.95%、99.98%、100.05%(RSD=0.46)。由此可見,米索硝唑pH敏感脂質體經預處理后,采用UV法和HPLC法所測得的主成分含量之間無明顯差異。
3 討論
在對制劑進行質量控制時,選擇合適的檢測方法是關鍵,既要準確、可靠,又要方便、快捷。通常,對有紫外吸收的成分,其含量測定的首選方法是UV法或HPLC法[11]。由于2015年版《中國藥典》未收錄米索硝唑及其制劑,因此,對米索硝唑及其制劑中主成分的含量測定方法沒有現行的標準。
參照文獻[12]報道,米索硝唑的最大吸收波長為320~325 nm。本試驗通過紫外光譜掃描確定米索硝唑最大吸收波長為322 nm,而其pH敏感脂質體中的輔料磷脂酰乙醇胺在此波長下有明顯干擾。磷脂分離方法包括有機溶劑萃取法、超臨界流體萃取法、乙酰化法、柱層析法、膜分離法、金屬離子沉淀法等[13],其中金屬離子沉淀法操作簡單,即通過重金屬離子與磷脂酰乙醇胺結構中的羧基或胺基強力結合,使其沉淀并可通過離心除去,不影響主成分的測定,更適合本研究。常用的金屬離子沉淀劑包括CoCl2、ZnCl2、NiCl2等。本課題組通過預試驗對比發現,ZnCl2反應條件溫和,不與主成分反應,不干擾主成分的紫外吸收,更適合本研究。采用ZnCl2預處理后的脂質體溶液中經HPLC法分析證實已無磷脂酰乙醇胺存在,從而消除了其對主成分含量測定的干擾。
綜上所述,UV法結合金屬離子沉淀法操作簡便、準確,精密度、穩定性、重復性好,可用于米索硝唑pH敏感脂質體中主成分的含量測定,其結果與HPLC法含量測定結果一致。
