系統性硬化癥并腎功能不全患者的臨床分析
2019-01-17 02:06:04許雪蓮孫熒宮笑微李榮濱宋麗萍劉微田微劉莎
中外醫療 2019年32期
關鍵詞:系統性
許雪蓮 孫熒 宮笑微 李榮濱 宋麗萍 劉微 田微 劉莎
[摘要] 目的 了解系統性硬化癥并腎功能不全患者的臨床表現及病理特點。方法 對2002年1月—2017年3月齊齊哈爾第一醫院風濕科病房和門診收治的36例臨床確診的系統性硬化癥并腎功能不全患者進行回顧性臨床分析,并對其中11例患者進行了腎組織病理學檢查。結果 在36例系統性硬化癥合并腎功能不全患者中,臨床表現為腎危象(SRC)16例,慢性腎損傷和炎癥性腎損害為13例,重疊結締組織病為3例,藥物相關為3例,其他1例。11例患者腎組織學顯示系統性硬化腎功能不全腎臟血管改變為多樣性,其中6例腎活檢示小葉間動脈和入球小動脈內皮細胞增生、腫脹、內膜粘液性水腫,腎病,另2例表現為藥物因素。結論 系統性硬化癥腎功能不全可能由疾病本身、重疊疾病、藥物等因素所致。疾病本身所致的腎損害可表現為腎危象、炎性改變或慢性損害。系統性硬化癥腎功能不全原因不清時可行腎臟活檢檢查,以幫助臨床治療及判斷預后。
[關鍵詞] 硬皮病,系統性;腎功能不全
[中圖分類號] R322.6+1? ? ? ? ? [文獻標識碼] A? ? ? ? ? [文章編號] 1674-0742(2019)11(b)-0007-03
[Abstract] Objective To understand the clinical manifestations and pathological features of patients with systemic sclerosis complicated with renal insufficiency. Methods A retrospective clinical analysis was performed on 36 patients with clinically confirmed systemic sclerosis and renal insufficiency admitted from January 2002 to March 2017, qiqihar first hospital rheumatology ward and outpatient department. Renal histopathology was performed in 11 of them. Results Among 36 patients with systemic sclerosis complicated with renal insufficiency, the clinical manifestations were 16 cases of renal crisis (SRC), 13 cases of chronic kidney injury and inflammatory renal damage, and 3 cases of overlapping connective tissue disease. 3 cases of drug-related, the other 1 case. Renal histology of 11 patients showed systemic sclerosing renal dysfunction and renal vascular changes were diverse, of which 6 renal biopsy showed interlobular artery and afferent arteriolar endothelial cell hyperplasia, swelling, intimal mucinous edema, nephropathy, and 2 case were manifested as a drug factor. Conclusion Systemic sclerosis may be caused by the disease itself, overlapping diseases, drugs and other factors. Kidney damage caused by the disease itself can manifest as a kidney crisis, an inflammatory change or a chronic damage. Systemic sclerosis Renal dysfunction is unclear when a renal biopsy is available to help with clinical treatment and prognosis.
[Key words] Scleroderma; Systemic; Renal insufficiency
系統性硬化癥(SSc)是一種少見病,難治病,主要發病年齡在30~50歲之間。女性發病率為男性的35倍[1-2]。在我國結締組織病中,硬皮病的發病率僅次于類風濕關節炎、系統性紅斑狼瘡,居于我國結締組織病發病的第3位。病因尚不完全清楚,臨床表現復雜,早期診斷困難,常不能早期治療,其腎臟受累尤為重要[3]。國內對SSc腎臟損害報道較少,行病理檢查的更是極少數,而對SSc合并腎功能不全的無報道。為此該文對2002年1月—2017年3月收治的36例確診的系統性硬化癥并腎功能不全患者進行了回顧性臨床分析,并對11例患者進行腎組織病理學檢查,以期探討其臨床意義,現報道如下。
1? 資料與方法
1.1? 病例納入
診斷標準:參照1980年美國風濕病學會(ACR)提出的系統性硬化癥診斷標準[4]。以及腎功能不全標準定為血肌酐≥132.5 μmol/L,尿素氮≥7.1 mmol/L或肌酐清除率<80 mL/min。
納入標準:①年齡:30~80歲;②符合診斷標準;③患者意識清楚。
排除標準:①有其他嚴重軀體疾病患者;②意識不清者;③依從性差,無法完成研究者。
收集該院病房和門診資料完整的系統性硬化癥并腎功能不全患者36例。
1.2? 研究方法
1.2.1? 臨床資料? 由病例記錄中獲得,收集患者年齡、性別、病程、血壓、血尿程度。
1.2.2? 生化指標檢測? 24 h尿蛋白定量,采用拜爾1650生化分析儀進行檢測。
血肌酐及尿素氮,采用TBA-40FR全自動生化分析儀,上海科華試劑,RANDOX控制品及校準品。
抗核抗體(ANA),采用免疫熒光法檢測。
抗ENA抗體,采用免疫印跡法(IBT)和酶聯免疫吸附試驗(ELISA),試劑由歐盟醫學診斷試劑公司提供。
ANCA,采用間接免疫熒光法(IIF)和ELISA,正常值為陰性,試劑由歐盟醫學診斷試劑公司提供。
1.2.3? 腎臟活檢? 標本分別進行常規光鏡(行HE、PAS、Masson及PASM染色)、免疫熒光及電鏡檢查。觀察電子致密物在系膜區、內膜下、基底膜內和上皮下沉積的情況。
1.3? 統計方法
應用SPSS 13.0統計學軟件進行分析數據,計數資料用[n(%)]表示,組間比較行χ2檢驗,P<0.05為差異有統計學意義。
2? 結果
2.1? 臨床特點
2.1.1? 基本情況? 36例患者中,女性29例,占81%,男性7例,占19%,男:女=1:4,發病年齡26~74歲,中位年齡為47.0歲,平均年齡(47.14±12.11)歲。20~30歲1例,占2.1%;30~40歲8例占22.2%;40~50歲14例,占38.9%,50~60歲及60~70歲均6例,各占16.7%,>70歲1例,占2.1%。在發病患者中30~40歲及40~50歲是發病的高峰年齡。既往病程4個月~29年,平均(75.50±88.34)月。在36例系統性硬化癥合并腎功能不全患者中,臨床表現為腎危象(SRC)16例,慢性腎損傷和炎癥性腎損害為13例,重疊結締組織病為3例,藥物相關為3例,其他1例。
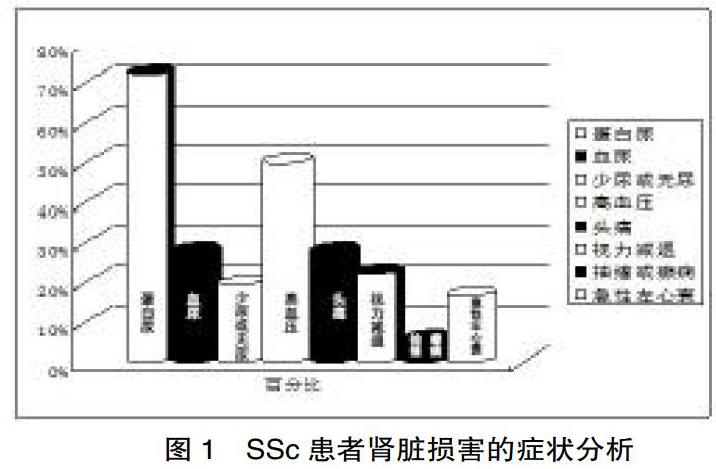
2.1.2? 臨床表現? 36例SSc并腎功能不全患者肌酐水平在171~1014 μmol/L,尿素氮明顯升高。出現蛋白尿26例,占72.2%;血尿10例,占27.8%;少尿或無尿7例,占19.9%,高血壓18例占50%;視力減退8例占22.2%,病情危重出現抽搐或癲癇的2例,占5.6%;出現急性左心衰的6例,占16.7%,見圖1。
2.2? 免疫學檢查結果分析
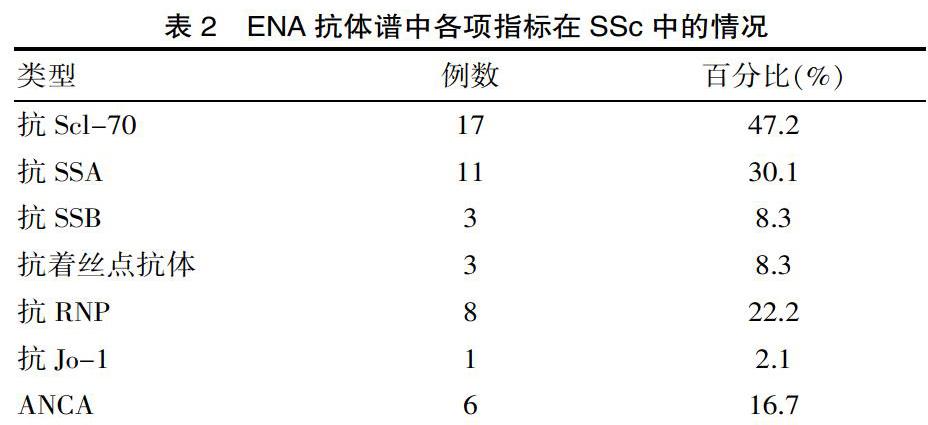
免疫學檢查結果分析具體數據見表2。
2.3? 腎臟病理表現
該組行腎活檢的11例患者中,6例光鏡下表現為惡性高血壓的腎臟損傷,受累血管為入球小動脈和小葉間動脈,血管損傷包括小動脈內皮細胞增生、腫脹,內膜粘液性水腫,內彈力纖維斷裂,動脈內膜呈同心圓纖維素性增厚,管腔狹窄,伴有腎小球彌漫性缺血,部分患者可小血管腔內附壁血栓形成,腎間質灶性單核細胞浸潤,間質纖維化,小管萎縮。
2例患者光鏡下可見腎小球新月體形成,大部分為大型纖維性新月體,余為細胞纖維性新月體,可見節段性系膜細胞和內皮細胞增生和系膜基質增多,部分毛細血管袢受壓變窄,部分腎小球基質膜變性、增厚、皺褶,可見節段性纖維素性壞死,腎小管基底膜增厚和腎小管萎縮,基質可見多灶性的纖維化,伴有大量單核細胞為主的炎細胞浸潤。
1例患者為慢性腎損害,其病理表現腎小球呈球性或基質硬化(其中2個伴有纖維性新月體,部分腎小球系膜區增寬,系膜基質增多,腎小球基底膜增寬,扭曲,毛細血管袢塌陷,部分腎小球擴張,可見蛋白管型,灶性小管萎縮, 浸潤劑間質纖維化,可見小葉間增厚及硬化。
2例患者的腎功能不全與服用藥物相關。1例為口服含有木通的中藥有關,另一例為應用青霉胺造成腎病綜合征。病理可見腎小球細胞數未見明顯增多,偶見節段性系膜細胞增生和系膜基質增多,毛細血管襻開放良好。GBM未見明顯增多,腎小管上皮細胞可見空泡變性,可見數灶性或小灶性輕度TBM增厚和腎小管萎縮。
2.4? 治療和預后
36例患者均接受了糖皮質激素治療,29例接受環磷酰胺治療,15例腎功能進展迅速,行血液透析治療,1例因咯血行血漿置換治療。SRC的16例患者均接受了ACEI和(或)ARB類藥物治療,劑量為常用劑量的3~4倍,患者血壓均控制滿意,腎功能均未恢復正常。8例患者最終因腎、心功能衰竭而死亡。
3? 討論
SSc主要表現為皮膚及內臟緩慢進展的炎癥、纖維化及萎縮等表現。它以膠原增生、炎癥細胞浸潤、血管阻塞、缺血萎縮、免疫異常等為特點。該病女性多發,好發年齡在30~50歲,20歲以下患者少見。該資料男女比例約為1:4,高發年齡為30~40歲及40~50歲,與Jaeger等[1]文獻報道相符。
該文36例患者病程中有雷諾現象32例(88.9%),患者最常見的首發癥狀為雷諾現象,判斷雷諾現象是原發的,還是SSc首發癥狀是很重要的問題,原發性雷諾現象患者大多數無血管結構的改變或組織缺血性損害,而皮膚毛細血管鏡檢查SSc患者的甲皺襞毛細血管,有報道[5-6]認為SSc患者的甲皺微循環變化嚴重程度與內臟器官受累相關,因此可間接反映內臟器官受累情況。該組病例另一常見的首發癥狀是手指腫脹/手指硬化,病程中100.00%患者出現手指腫脹/手指硬化,可見,手指腫脹/手指硬化、雷諾現象是提示SSc的最重要臨床表現。
對于該病的治療,36例患者均應用糖皮質激素治療,根據病情調整激素劑量。在SSc的治療中,激素的使用可減輕皮膚炎性水腫、減輕肺動脈高壓,但大劑量應用可能增加SRC患者腎臟危象發生的危險性。SRC的16例患者均接受了ACEI和(或)ARB類藥物治療,劑量為常用劑量的3~4倍[7-8]。ACEI藥物的應用在治療SRC上已經達成共識。其能明顯升高SRC患者的生存率(未用ACEI者12個月生存率僅為24%,而應用者達85%[9-10]。血壓的控制應該循序漸進[11-12],目標是每24小時使收縮壓下降10~20 mmHg,直至血壓降至正常范圍。
綜上所述,系統性硬化癥并腎功能不全的病因病機、臨床表現、治療方案等都不盡相同,應根據不同原因制定不同的治療方案,必要時行腎穿病理檢查加以鑒別。
[參考文獻]
[1]? Jaeger VK, Wirz EG,Allanore Y,et al.Incidence and predi ctors of cutaneous manifestations during the early course of systemic sclerosis-A 10 year longitudinal study from the eustar database.Ann[J].Rheum.Dis,2016(75):1285-1292.
[2]? Gordon JK,Domsic RT.Clinical Trial Design Issues in Systemic Sclerosis:an Update.Curr[J].Rheumatol,Rep,2016(18): 38.
[3]? Kumar S, Singh J, Mendoza F, et al.133 Role of Muscarinic-3 Receptor (M3-R) in Pathogenesis of Gastrointestinal Dysmotility in Systemic Sclerosis:Correlation With Disease Duration and Effects of IVIG[J].Gastroenterology, 2016, 150 (4) :S32.
[4]? Dimitroulas, T Daoussis, Garyfallos A,et al. Molecular and Cellular Pathways as Treatment Targets for Biologic Therapies in Systemic Sclerosis[J]. Curr Med Chem,2015(22):1943-1955.
[5]? Korman? BD,Criswell LA.Recent? advances? in? the genetics of systemic sclerosis: toward biological and clinical significance[J]. Curr Rheumatol Rep, 2015(17): 21.
[6]? 張嘉倩,屠文震.124例系統性硬化癥患者臨床特點分析[J].風濕病與關節炎,2018,7(1):35-39.
[7]? Baird, K Comis, LEJoe, et al. Imatinib Mesylate? for? the? Treatment? of? Steroid-Refractory Sclerotic-Type Cutaneous Chronic Graft-versus-Host Disease[J].Blood Marrow Trans plant, 2015( 21): 1083–1090.
[8]? Yanaba K. Strategy for treatment of fibrosis in systemic scler osis: Present and future. J[J].Dermatol, 2016( 43): 46-55.
[9]? Desbois AC,Cacoub P.Systemic sclerosis:An update in 2016. Autoimmun[J].Rev,2016(15):417-426.
[10]? Murdaca, G.;Contatore, M.;Gulli, R.;Mandich, P.;Puppo, F.Genetic factors and systemic sclerosis.Autoimmun[J].Rev,2016(15): 427-432.
[11]? Yanaba, K.Strategy for treatment of fibrosis in systemic sclerosis:Present and future[J].Dermatol,2016(43):46-55.
[12]? Gordon JK,Domsic RT.Clinical Trial Design Issues in Systemic Sclerosis:an Update.Curr[J].Rheumatol,Rep,2016(18): 38.
(收稿日期:2019-08-16)
猜你喜歡
臨床誤診誤治(2021年12期)2021-12-04 00:25:45
臨床誤診誤治(2021年12期)2021-12-04 00:25:45
中國新聞周刊(2021年9期)2021-03-29 20:33:56
中學歷史教學(2016年10期)2017-01-15 14:26:09
中國男科學雜志(2016年5期)2016-12-01 05:20:21
鄭州大學學報(醫學版)(2015年1期)2015-02-27 14:50:31
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
現代檢驗醫學雜志(2015年6期)2015-02-06 01:44:03
西南軍醫(2015年5期)2015-01-23 01:25:06
名作欣賞(2014年29期)2014-02-28 11:24:31
