一種不易誘發耐藥性的新型抗生素teixobactin的研究進展
2019-01-30 06:49:02張煥云李瑞娟
中國抗生素雜志 2019年1期
張煥云 李瑞娟
(山東大學微生物技術國家重點實驗室,山東大學-亥姆霍茲生物技術研究所,青島 266237)
近些年,隨著抗生素的廣泛使用甚至濫用,細菌耐藥性逐漸增加,耐藥性細菌感染不僅嚴重危害人類的健康,而且已經成為世界范圍的棘手問題。因此,迫切需要挖掘作用于獨特分子靶標的新型抗生素[1-2]。
2015年,美國藥物學家Kim Lewis通過iChip(isolation chip)技術[3]在一株難培養的土壤微生物Eleftheria terrae中發現的新型抗生素teixobactin對多種革蘭陽性致病菌,包括很多臨床耐藥菌具有顯著的抑制活性,并且不易誘發耐藥性[4]。Teixobactin能夠分別和細菌細胞壁上肽聚糖前體脂質II和壁磷壁酸前體脂質III的保守序列結合,而這兩個靶點是非常難通過突變而發展耐藥性的[5]。這使teixobactin作為一種對抗耐藥性致病菌的先導化合物顯示出巨大的潛力[6-7]。本篇綜述總結了自teixobactin發現以來的研究進展,重點介紹了其不易誘發耐藥性的作用機制,并對其未來在生物合成方面的研究進行了展望。
1 Teixobactin的化學結構和生物活性
Teixobactin是由11個氨基酸殘基組成的環縮肽,包含6個天然L-氨基酸,1個非天然氨基酸(L-alloenduracididine,L-allo-End10),N-Me-D-Phe和3個其他的D-氨基酸。C端的D-Thr8和L-Ile11通過酯鍵形成十三元環的四肽內酯亞結構(圖1)。Teixobactin是細菌為了自我防御而產生的,必然經過了長期的自然進化和選擇來形成最優化的結構,其顯著的抑菌活性取決于這種獨特的分子結構[8]。
1.1 L-allo-End—teixobactin中的非天然氨基酸
L-allo-End是一個含有五元結構環胍基團的非天然氨基酸。L-End是L-allo-End的非對映異構體,已被發現存在于多種天然產物中,比如甘露霉素(mannopeptimycin)[9]和恩拉霉素(enduracidins)[10],并對于維持其生物活性具有重要的作用。同位素標記實驗證明L-End是由L-Arg產生的[11]。比較分析甘露霉素和恩拉霉素的生物合成基因簇,發現3對高度同源的酶:EndP/MppP(80%),EndQ/MppQ(68%)和EndR/MppR(75%)[12]。生化和結構研究表明,L-Arg經過PLP依賴的脫氨羥化酶MppP的催化生成2-羰基-4-羥基-5-胍基戊酸[13],后經脫羧酶MppR和PLP依賴的轉氨酶MppQ催化生成L-End[14]。盡管L-allo-End和L-End在C4處有不同的立體化學構型,L-allo-End在teixobactin中的生物合成可能也遵循與L-End類似的途徑。然而,在teixobactin的生物合成基因簇中并沒有發現L-allo-End生物合成所需的同源基因。這可能是因為L-allo-End的生物合成基因簇位于E. terrae基因組的其他位置。這一假設還有待研究和考證。
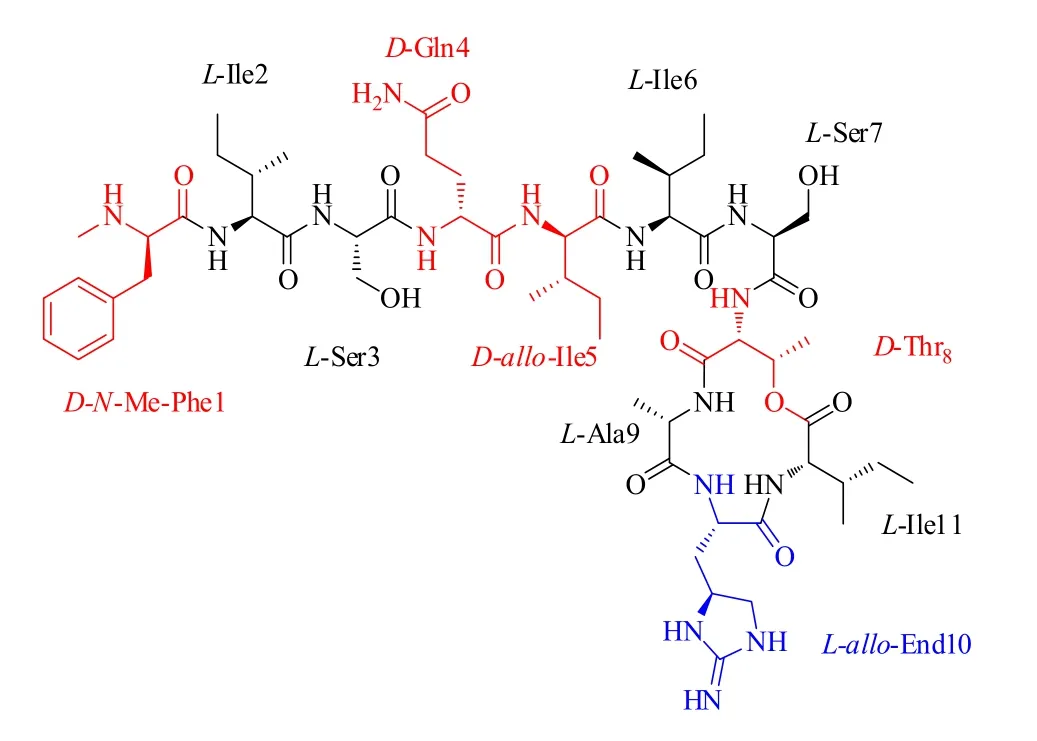
圖1 Teixobactin的化學結構Fig.1 Chemical structure of teixobactin.
L-allo-End的化學合成過程非常復雜。目前該氨基酸在市場上購買不到,因此成為了teixobactin化學合成的一大挑戰。Rudolph課題組[15]首次報道了L-allo-End的化學合成,L-End作為一個副產物,與L-allo-End的產量比率是1:6。2015年,Craig課題組[16]報道了高度立體選擇性地合成L-allo-End的新方法。從Boc-保護的反式-羥基脯氨酸中通過10步反應提供L-allo-End,具有超過50:1的非對映立體選擇性。Payne課題組[17]報道了另一種方法來合成L-allo-End,以保護的L-Asp酯作為起始材料。在這個合成過程中,采用三仲丁基硼氫化鋰的立體選擇性酮還原反應來引入立體化學(dr: 5:1 (2S,4R):(2S,4S)),通過快速柱色譜除去次要非對映異構體產物。通過使用類似的策略,Reddy課題組[18]實現了L-allo-End的克級合成,足以用于teixobactin的化學全合成研究。
1.2 Teixobactin的生物活性
體外抑菌活性研究表明,teixobactin對大多數革蘭陽性菌都具有顯著的抑制活性,尤其是對梭狀芽胞桿菌和炭疽桿菌的最低抑制濃度(minimal inhibitory concentration, MIC)達到了納克級[4]。Teixobactin對臨床上治療周期長且成本高的結核桿菌也有很強的抑制活性[4]。對于多種耐藥菌如MRSA和耐萬古霉素腸球菌(VRE)等,teixobactin都具有顯著的抑制活性,其MIC均在1μg/mL以下。Teixobactin不能有效抵抗大多數革蘭陰性菌,但是對外膜通透性屏障缺陷的E. coliasmB1表現出良好的活性[4](表1)。體外毒理學實驗研究表明,teixobactin沒有明顯的毒副作用[4]。

表1 Teixobactin對致病微生物的抑制活性[4]Tab. 1 Activity of teixobactin against pathogenic microorganisms[4]
體內活性研究顯示,teixobactin在體內穩定存在,且毒性低。在小鼠MRSA感染敗血癥模型的研究中,teixobactin的半數保護量(PD50)為0.2mg/kg,低于臨床治療MRSA感染的萬古霉素(2.75mg/kg)劑量[4]。Teixobactin的體內藥動學結果也令人非常滿意,小鼠接受20mg/kg的teixobactin,其血藥濃度維持在MIC值以上的時間可達4h[4]。
2 Teixobactin的作用機制
大量實驗研究表明,teixobactin不易誘發細菌產生耐藥性。即使在4倍MIC劑量的teixobactin鋪板時,也沒能得到耐teixobactin的金黃色葡萄球菌和結核分枝桿菌[4]。在teixobactin的亞最低抑制濃度(sub-MIC)下連續傳代27d,同樣沒有發現金黃色葡萄球菌的耐藥性突變株[4]。因此,teixobactin作為對抗耐藥性致病菌的先導化合物顯示出巨大的潛力。這主要取決于teixobactin多靶標作用于細胞壁的特殊機制。
2.1 高度保守的肽聚糖前體—脂質II(LII)
脂質II(LII)是細菌細胞壁生物合成的最重要的中間產物之一。因其結構具有高度保守性,數十年來被公認為抗生素的理想靶標[19]。從結構上講,一個LII分子由一個包埋在細胞膜中的細菌萜醇烴鏈(C55)、十一異戊烯基-N-乙酰氨基葡萄糖(UDPMurNAc)和N-乙酰胞壁酸(GlcNAc)的二糖、附著在MurNAc上的五肽(通常帶有L-Ala-γ-D-Glu-L-DAP-DAla-D-Ala的序列)和連接細菌萜醇(C55)錨和MurNAc的焦磷酸鹽基團(PP)組成。二糖和五肽形成的肽聚糖亞基進一步通過五肽相互交聯,提供細胞壁的機械強度。該分子首先裝配到細胞質側的細菌萜醇(C55)錨上,然后易位至五肽連接的二糖所在的周質側,從錨上釋放出來構建細胞壁。剩余的C55-PP通過機制不明確的回收途徑轉移回細胞質側。因此,阻止LII移位是通過干擾細胞壁合成途徑中的關鍵步驟來殺滅細菌的一種策略。由于LII的高度保守性,細菌對于作用于LII的抗生素產生耐藥性的幾率非常低。
萬古霉素和LII之間的相互作用已經被充分研究。萬古霉素通過五肽的D-Ala-D-Ala片段上的5個氫鍵結合至LII,形成復合物[20]。這些復合物中的幾個甚至可以通過D-Ala-D-Ala片段形成超級復合物[21]。然而,經過20年的進化和選擇,細菌通過突變D-Ala-D-Ala的五肽末端為D-Ala-D-Lac從而對萬古霉素產生了耐藥性。萬古霉素對突變的LII的結合親和力降低了1000倍左右,因而大大降低了萬古霉素的抑菌效果,產生耐藥菌株[22]。尼生素(nisin)能有效抑制引起食品腐敗的革蘭陽性菌。它作用于LII中高度保守的焦磷酸基團(PP),目前沒有發現耐藥性[23]。馬拉西汀(malacidins)是最近發現的可對抗多種耐藥菌的鈣依賴性抗生素,研究表明它也不易誘發細菌產生耐藥性[24]。用馬拉西汀處理金黃色葡萄球菌,細胞壁中積累UDP-MurNAc-五肽,說明馬拉西汀作用的靶標也是LII。
2.2 壁磷壁酸(WTA)前體—脂質III(LIII)
磷壁酸是革蘭陽性菌細胞壁所特有的成分。根據其在菌體上的分布,磷壁酸可分為2類,一類是壁磷壁酸,其末端以磷酸二酯鍵錨定到肽聚糖上;另一類是脂磷壁酸,錨定到細菌細胞膜上。抑制壁磷壁酸的生物合成對于細菌來說是致命性的,因為在細菌體內積累有毒中間體[25]。另外,壁磷壁酸可以固定自溶素,防止肽聚糖被水解。抑制壁磷壁酸生物合成有助于細菌釋放自溶素,使細胞溶解,進而死亡[26]。
2.3 對抗耐藥性—teixobactin獨特的雙重作用方式
Teixobactin可以雙重靶向LII和LIII[4]。實驗證明,teixobactin可以強效地抑制肽聚糖的生物合成,對于DNA、RNA和蛋白質均沒有實質性作用[4]。添加純化的LII可以阻止teixobactin抑制細菌生長的作用,說明teixobactin直接作用于LII[4]。化學計量分析表明teixobactin以2:1的比率結合LII[5]。進一步分析表明,teixobactin結合LII中高度保守的焦磷酸鹽基團(PP)和MurNAc基團。并且teixobactin還能夠有效地結合LIII中的UDP-PP-GlcNAc,有效地抑制壁磷壁酸的生物合成[4]。Teixobactin具有強有效的抑菌活性和無耐藥性潛力,得益于它的雙重作用方式,即同時抑制肽聚糖前體LII和壁磷壁酸前體LIII的生物合成(圖2)。二者通過協同效應,導致細胞壁損傷,胞壁離域,蛋白酶自溶,隨后細胞溶解[5]。這種高保守的雙重靶標(LII和LIII)作用機制使致病菌幾乎不可能對其產生耐藥性。
最近,teixobactin-LII復合體的結構模型已經被闡明,發現了4種不同的結合模式(BM1、BM2、BM3和BM4)[27]。同時,結果表明,teixobactin的環狀結構域對于識別LII具有重要作用[27]。這些新發現為進一步理解teixobactin與LII的作用機制提供了新思路。
3 Teixobactin及其類似物的化學合成
3.1 Teixobactin的化學全合成
成功地高效合成L-allo-End克服了teixobactin合成的主要障礙。目前為止,兩個科研團隊采用了固相多肽合成方法(SPPS),通過截然不同的合成途徑完成了teixobactin的全合成[17,29]。
悉尼大學Payne課題組[17]利用Fmoc-SPPS首次實現了teixobactin的全合成。首先,他們先合成了可以直接安裝入Fmoc-SPPS的經適當保護的L-allo-End,即FmocEnd(Cbz)2-OH;然后組裝teixobactin的縮酚酞鏈;在弱酸性條件下裂解樹脂,隨后液相環化;然后在強酸性條件下,總體側鏈去保護,包括L-allo-End的Cbz保護;最后產出teixobactin。通過24步過程,可以達到3.3%的總收率[17]。
香港大學李學臣課題組[29]報道了一種基于Ser/Thr連接的縮合反應來合成teixobactin。無保護肽段的N-Ser或N-Thr殘基和另一個無保護肽段的C-水楊醛酯之間的化學選擇性反應產生一種N, O-芐叉縮醛鍵合產物。它酸解時,在連接位置產生天然的肽鍵。含有N-Ser殘基的環肽通過Fmoc-SPPS合成,含有C-水楊醛酯的線性肽片段通過Boc-SPPS合成,通過Ile6和Ser7的高效連接來合并線性的六肽和環化的五肽,從而合成teixobactin,縮合產物經HPLC純化后收率為37%。值得注意的是,Arg10-teixobactin和Orn10-teixobactin的合成比teixobactin更順利。這表明,天然形態的teixobactin的合成很可能會遇到一系列與類似物合成截然不同的挑戰。
3.2 Teixobactin類似物的化學合成及其構效關系
L-Arg10-teixobactin是第一個報道的teixobactin類似物,報道時間比teixobactin的全合成還要早[30-31]。它常作為研究teixobactin構效關系的模型。目前,對于teixobactin構效關系的研究,全部基于相對容易合成的teixobactin類似物[8,30-43]。表2列舉了目前成功合成的具有抑菌活性的teixobactin類似物。
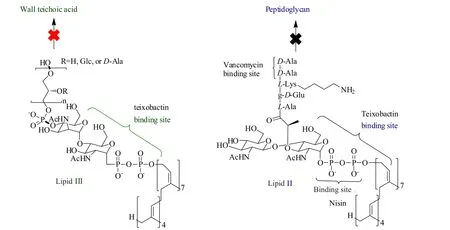
圖2 Lipid II和Lipid III的結構示意圖以及teixobactin的作用靶點[28]Fig.2 Structures of Lipid II and Lipid III with the targets of teixobactin[28]
與天然的teixobactin相比,L-Arg10-teixobactin的抑菌活性有所降低,因此L-allo-End是維持teixobactin最佳生物活性的關鍵殘基[30-31]。胍基對于維持活性是非必需的,因為Lys10-teixobactin和Orn10-teixobactin針對各種葡萄球菌的抑菌活性比Arg10-teixobactin更強,其MIC值在萬古霉素(0.5μg/mL)范圍內[32-33]。進一步的構效關系研究揭示了10位氨基酸殘基的絕對立體化學不是必要的。而整個環狀結構域的相對立體化學是非常關鍵的[32]。利用L-allo-End的電子等排體如Lys、Orn、L-2,4-Diaminobutyric acid (DAB)和L-1,3-diaminopropionic acid(DAP)[34]或疏水基團如Ile或Leu[35]L-allo-End10的類似物表現出出色的抑菌活性,證明L-allo-End或許并不是不可替代的。這一結論需要更進一步的體內外活性研究以及藥效學研究。
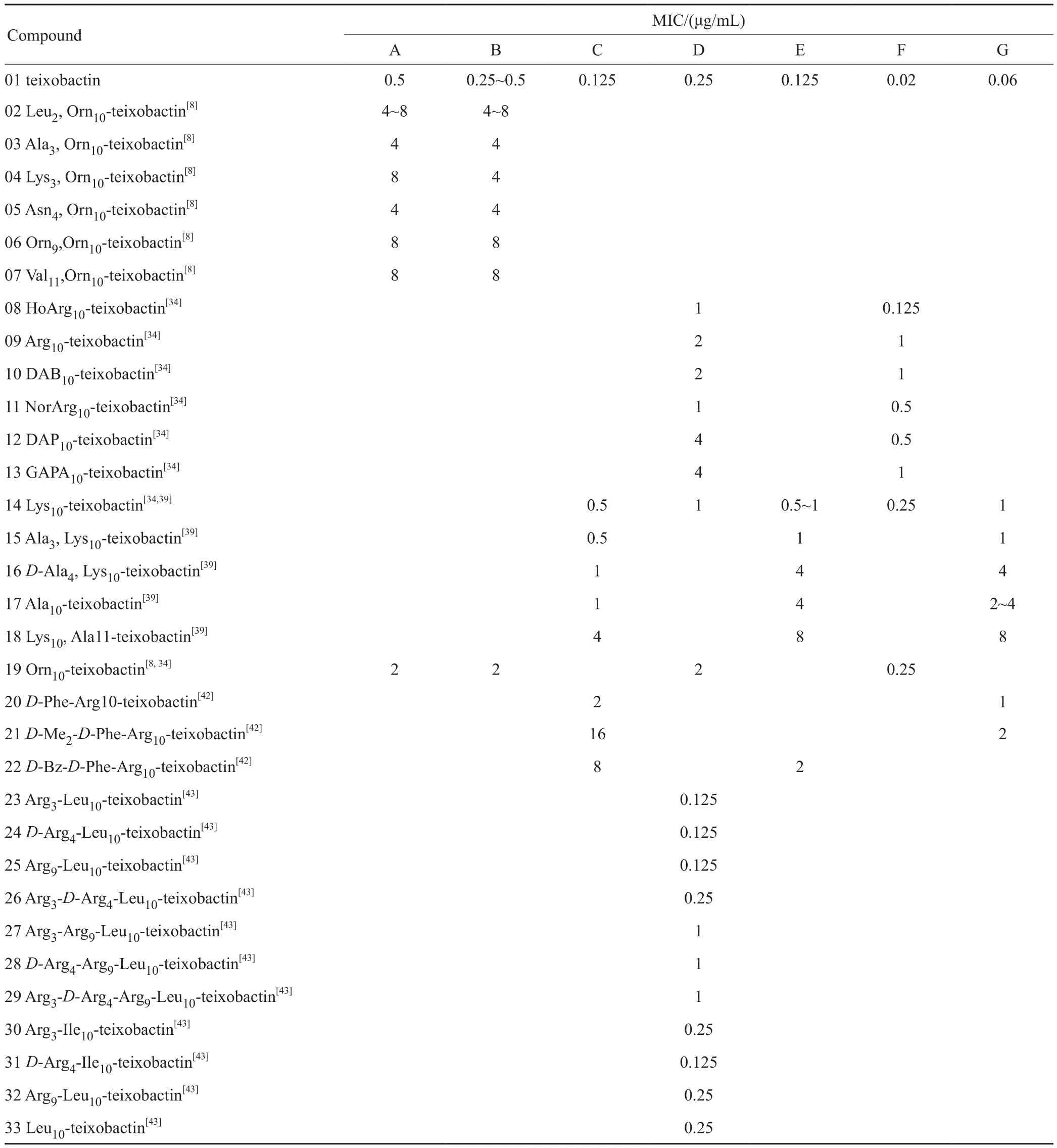
表2 人工合成的teixobactin類似物及其抑菌活性Tab. 2 Synthesized teixobactin analogues with relatively good antibacterial activity
D-氨基酸對活性也起重要作用。當teixobactin結構中的4個D-氨基酸殘基,即:NMe-D-Phe1、D-Gln4、D-allo-Ile5和D-Thr8全部或者逐個被L-氨基酸取代時,抑菌活性幾乎全都喪失了[36-37]。Lys/Ala掃描(Lys/Ala scanning),即按順序將每個殘基替代為Lys/Ala,以研究疏水性與親水性的平衡對于化合物保持抗菌活性的重要性。對Arg10-teixobactin進行Lys掃描[38]和對Lys10-teixobactin進行Ala掃描[39]的結果共同表明,疏水的Phe1、Ile2、Ile5、Ile6和Ile11對于保持活性非常重要,而極性不帶電的Ser3、Gln4和中性的Ala9具有良好的耐受性。值得注意的是,Ala取代不帶電的Ser7的類似物的抗菌活性顯著下降,說明Ser7對于保持活性是至關重要的[39]。隨后的晶體結構證明,Ala9的NH基團與Ser7的側鏈形成了氫鍵[40]。進一步研究表明,維持teixobactin活性所需的最大正電荷數是3~4個,引入額外的正電荷將導致活性的完全損失[41]。另外,teixobactin的N-Me-Phe1對于保持活性非常重要,對其酰基化或烷基化都會導致活性完全喪失[42]。
Teixobactin類似物是否可以發展成為臨床應用的新的抗菌藥物,將化合物分子從發現階段轉換為臨床使用的抗生素需要面對眾多挑戰,如針對廣泛病原體高效的活性和無毒副作用之間的平衡,以及親水性和疏水性之間的平衡以解決水溶性問題等。雖然Leu10-teixobactin和Ile10-teixobactin具有顯著的抑菌活性,但是其疏水性的增加不利于其水溶性。將Ser3、Gln4和Ala9替換為陽離子的Arg殘基可以模擬天然teixobactin的疏水性與親水性之間的平衡。通過這種方式,帶有兩個陽離子殘基的類似物,如D-Arg4-Leu10-Teixobactin實現了與teixobactin類似的疏水性和親水性之間的最佳平衡,同時具有與teixobactin一致的抑菌活性[43]。體內毒理學研究和小鼠細菌性角膜炎模型研究均證明D-Arg4-Leu10-Teixobactin無細胞毒性作用[43]。這項工作在合成安全高效的簡化的teixobactin類似物方面取得了重大進展,推動了新型對抗耐藥性菌株的抗菌藥物的研發進程。
4 Teixobactin的生物合成
生物信息學分析發現了teixobactin的生物合成基因簇(GenBank accession number KP006601),其包括兩個NRPS編碼基因,txo1和txo2。這兩個NRPS編碼基因共含有11個模塊(module),計算機預測的每個腺苷酰結構域(A)的底物特異性都與teixobactin的結構相匹配[4]。因此,teixobactin遵循NRPS典型的共線性規則,其中每個模塊負責整合新生肽鏈中的一個特定的氨基酸(圖3)。通過L-Ile11與D-Thr8的內酯化反應將線性肽鏈從NRPS組裝生產線上釋放。甲基轉移酶(MT)結構域存在于第1模塊中,其負責D-Phe1的N-甲基化。值得注意的是,txo2包含兩個串聯硫酯酶(TE)結構域,這種結構在NRPS中是很罕見的。研究證明,兩個串聯硫酯酶在功能上是可互換的,并且很有可能協同作用,代表了一種前所未有的NRPS酶學卸載機制[44]。另一點值得提出的是,模塊8包含一個額外的縮合(C)結構域,它的功能還有待研究。一些基因與txo1和txo2相鄰,它們的功能尚不清楚,這些基因可能涉及調控、產物產出等。
近年來,本團隊的RecET同源重組技術取得了重要進展,極大地促進了微生物生物合成基因簇組裝、克隆和異源表達的研究[45-48]。這項技術是依托λ或Rac噬菌體的內源性DNA重組蛋白(Redα/β及RecE/T)而建立起來的一套完善的基因重組技術體系。其在大片段基因簇方面的有效應用,可以實現大片段、高重復的基因簇的載體化、工程化連接。近期,利用RecET同源重組技術,本團隊完成了teixobactin生物合成基因簇(約54kb)的人工合成和組裝,期望通過生物合成和異源表達的方式,更為高效、便利地大規模生產teixobactin及其類似物。
5 展望
自2015年teixobactin發現以來,科研學者們開展了大量的研究,包括作用機制、化學全合成、構效關系等。在此期間,先后使用和發展了多種高效的化學方法用于teixobactin及其類似物的合成,合成獲得的化合物達上百種。顯然,這些突破性成果是一個激動人心的開端。Teixobactin顯著的抑菌活性、不易誘發耐藥性的潛力及其對哺乳動物細胞的低毒作用等使其成為一種巨大開發前景的天然藥物。
目前已經成功合成了與teixobactin抑菌活性一致的類似物,這項成果向合成安全高效的簡化的teixobactin類似物的方向邁進了一大步,但是更進一步的構效關系研究以及毒理學和藥理學研究也是必不可少的。另外,目前對于teixobactin與它的靶點之間的具體的作用方式還不是很清楚,這方面的繼續研究對于理解teixobactin強有效的抑菌活性以及應對將來可能的耐藥性發展意義重大。
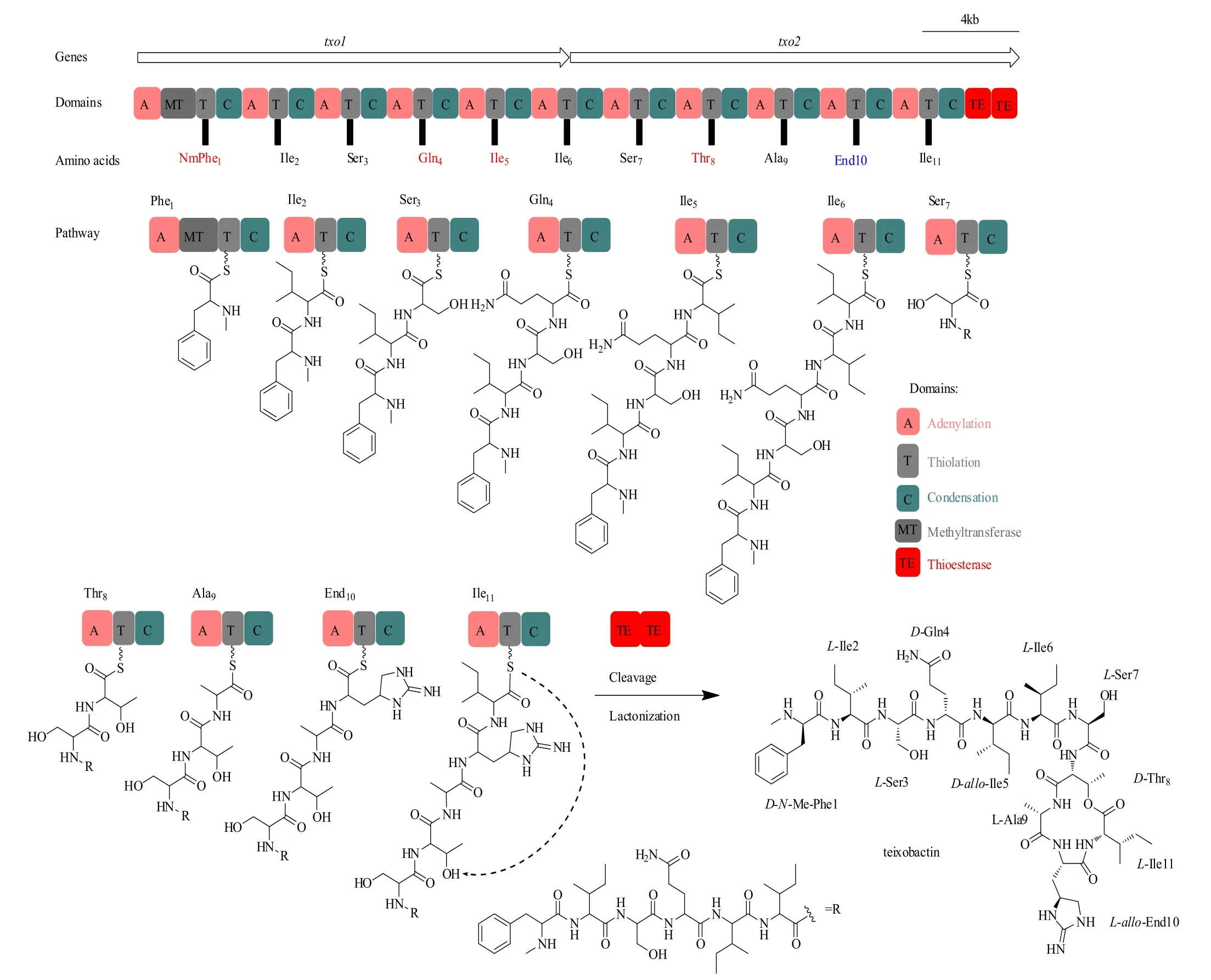
圖3 預測的teixobactin生物合成途徑[4]Fig.3 Predicted teixobactin biosynthetic pathway [4]
未來研究中另外一個重要的方面是teixobactin的生物合成,這在很大程度上滯后于化學合成研究。Teixobactin在野生菌中具體的生物合成途徑以及后修飾是怎樣的;L-allo-End在野生菌中的生物合成是如何實現的;如何提高teixobactin的總體產量,相信這方面的研究仍任重道遠。RecET同源重組技術建立了對大型生物合成途徑進行高通量的克隆、組裝、修飾及異源表達的技術平臺,首次實現了teixobactin基因簇的體外組裝,為teixobactin基因組功能研究提供了極大的便利。
已知的天然產物僅僅代表自然界全部的天然產物生物合成能力的冰山一角,不可培養的微生物研究具有巨大的前景。大多數微生物有待挖掘,或者只是通過其基因組分析特征,至今還沒有培養出來。隨著微生物學和分子生物學技術的迅猛發展,挖掘這些尚未開發的新型抗生素對感染治療具有重大意義。
猜你喜歡
天天愛科學(2022年9期)2022-09-15 01:12:54
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科學大眾(2021年9期)2021-07-16 07:02:54
軍事文摘(2020年20期)2020-11-28 11:42:50
航空世界(2020年10期)2020-01-19 14:36:20
科技傳播(2019年22期)2020-01-14 03:06:54
