腫瘤中ERG基因的表達及其臨床研究進展
2019-03-07 11:31:40王文君於宇崔久嵬
天津醫藥 2019年2期
關鍵詞:融合
王文君,於宇,崔久嵬
ERG基因(ETS-related gene)位于21號染色體q22(21q.22.2)上,屬于 E26轉化特異性(E26 transformation-specific,EST)轉錄因子家族。ERG基因參與生理性和病理性血管生成、調節血管發育、控制內皮分化和重編程、維持外周血小板數量、參與正常巨核細胞(megakaryocytes,MEG)生成等[1]。近年來越來越多的研究發現,ERG基因在一些腫瘤中過表達,在腫瘤發生發展中發揮重要作用。ERG與其他基因易位產生融合基因產物可導致ERG過表達,進而調控ERG的靶基因和下游信號通路,參與腫瘤發生發展,例如前列腺癌(prostate cancer,PCa)、尤文肉瘤(Ewing′s sarcoma,EWS)和白血病等[2]。在白血病中,ERG本身作為癌基因也可以發生過表達。但是ERG在腫瘤中的具體作用機制尚未完全明確[3]。目前,關于ERG在腫瘤中的診斷、靶向治療和預后成為研究熱點,為腫瘤治療提供了新的方向。本文就ERG基因的結構功能、與腫瘤的關系以及靶向治療做一綜述。
1 ERG基因的結構
ERG基因跨越282個千堿基對(kilobase,kb),含有至少12個外顯子和3個近端啟動子(PⅠ-Ⅲ)。轉錄起始下游85 kb的區域已被鑒定為ERG增強子。在正常造血過程中該增強子具有活性,ERG通過其正向調節自身表達[1]。ERG的C′端有一個高度保守的ETS DNA結合結構域,可以識別含有核心GGA(A/T)的DNA序列,介導DNA結合和轉錄激活。N′端含有蛋白激酶C和PNT結構域的磷酸化位點,PNT結構域可形成螺旋-環-螺旋結構,供蛋白質-蛋白質相互作用和同/異二聚化[1],見圖1。
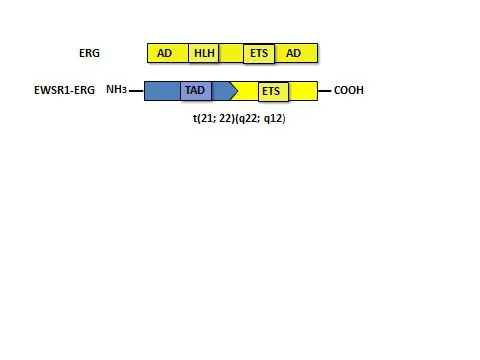
Fig.1 Diagram of translocation of ETS-related gene as well as EWSBR1 in ERG and Ewing′s sarcoma圖1 ERG和尤文肉瘤中EWSBR1和ERG易位的示意圖
2 ERG基因的正常表達與功能
正常情況下,ERG表達僅限于在內皮細胞(endothelial cell,EC)、MEG和軟骨細胞中。在ETS家族中,ERG在EC中表達最高,幾乎所有EC特異性表達的基因在其啟動子中都具有ETS結合位點[1]。在胚胎發育期間和出生后,ERG在胚胎發育、血管生成、造血系統、細胞凋亡等方面發揮重要作用。
在胚胎發育方面,ERG通過結合心內膜間質轉化(endocardial-mesenchymal transformation,EnMT)調控因子的啟動子和內含子來上調鋅指轉錄因子Snail家族表達,介導EnMT。EnMT減少而導致ERG突變體胚胎的心內膜墊形成缺陷,無法發育為成熟的功能瓣葉。
ERG還可促進血管成熟、穩定性和連接的完整性[1]。ERG通過調節多個EC基因的表達,如血管生成素-1、claudin-5(CLDN5)、細胞間黏附分子 2(intercellular adhesion molecule 2,ICAM-2)等[1]。在造血系統中,ERG是正常血液干細胞發育所需的ETS家族轉錄因子。ERG通過誘導造血干細胞(hematopoietic stem cell,HSC)自我更新因子的幾種基因表達,抑制HSC中原癌基因MYC驅動的HSC分化,從而調節HSC自我更新和分化之間的平衡。ERG還參與生成正常MEG、維持外周血小板(platelet,PLT)數量。ERG屬于造血干細胞和祖細胞(hematopoietic stem and progenitor cells,HSPC)表達的轉錄因子七肽(包括 GATA2、LYL1、TAL1、ERG、FLI1、RUNX1和LMO2)。
在成熟的脈管系統中,ERG促進穩態基因表達、抑制炎癥相關基因表達,如ICAM-1、纖溶酶原激活物抑制劑等,抑制細胞因子誘導的EC激活。在細胞凋亡方面,ETS轉錄因子參與各種細胞類型的細胞凋亡,并且通常作為保護性因子。最后,ERG是滑膜關節形成機制的重要組成部分,可以調節大多數滑膜細胞的發育行為。
3 ERG基因在腫瘤中的表達與臨床意義
ERG的表達具有組織特異性,作為譜系決定轉錄因子,ERG的過表達與腫瘤發病機制有關,包括促進腫瘤細胞增殖和腫瘤血管生成兩方面。
在腫瘤細胞增殖方面,在細胞分裂發生染色體易位過程中,ERG與其他基因發生易位,產生融合基因產物導致ERG過表達,例如PCa中雄激素調節跨膜絲氨酸蛋白酶(transmembrane protease,serine 2,TMPRSS2)與ERG融合形成TMPRSS2-ERG融合基因,EWS中的Ewing肉瘤斷裂區域1(Ewing sarcoma breakpoint region 1,EWSR1)基因和FUS RNA結合蛋白(FUS RNA binding protein,FUS)基因分別與ERG融合形成EWSR1-ERG和FUS-ERG融合基因,白血病中也形成FUS-ERG融合基因。另外,一些白血病中ERG作為癌基因發生過表達。ERG過表達后調控ERG靶基因和下游相關信號通路,發揮促腫瘤作用。
ERG還在腫瘤血管生成中發揮重要作用,血管生成是腫瘤生長和侵襲轉移的重要因素。Strzepek等[4]發現,相比ERG陰性PCa,ERG陽性PCa中有更高的微血管密度,而微血管密度是許多腫瘤的預后因素。腫瘤血管生成是由于血管生成因子和抑制因子之間的失衡,ERG誘導Notch配體DLL4和其他血管生成基因表達,在促進腫瘤新血管生成方面有重要作用[1]。血管內皮生長因子(vascular endothelial growth factor,VEGF)/絲分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)/細胞外信號調節激酶(extracellular signal-regulated kinase,ERK)信號傳導通路控制ERG活性,ERG磷酸化后招募p300至VEGF依賴性增強子來調節許多血管生成基因表達,即組成VEGF/ERK/ERG/p300轉錄途徑[5]。而靶向ERG磷酸化,選擇性阻斷血管生成和維持血管穩定性,可能利于降低ERG的致癌活性[5]。
3.1 ERG在PCa中的表達及臨床研究進展 PCa是全球癌癥死亡的第五大常見原因,發病機制尚不清楚,且治療方法局限,且患者易發展為去勢抵抗性前列腺癌,預后差[6]。2005年,Tomlins等[7]首次發現PCa中存在一種復發性基因組重排,即TMPRSS2和ERG基因之間發生融合,在高加索人、美國人中發生率為40%~60%,在激素難治性PCa中達50%~70%,在PCa的發生發展中有重要臨床意義。
3.1.1 PCa中的TMPRSS2-ERG融合基因及作用機制 前列腺上皮細胞通常不表達ERG,而基因融合導致ERG成為PCa中最常見的過表達原癌基因,并且驅動前列腺上皮內瘤變向PCa轉變。在PCa中,TMPRSS2的5′端非翻譯區和ETS家族基因[包括ETS 變體 1(ETV1),ETV4 和 ETV5]發生融合[7-8]。ERG和TMPRSS2是21號染色體長臂上相距3 Mb的2個基因,并且ERG內含子2以及TMPRSS2內含子1和2中均存在脆弱的位點和斷點。
ERG和TMPRSS2由于染色體易位或基因間片段缺失而融合形成TMPRSS2-ERG融合基因。雄激素信號募集雄激素受體(androgen receptor,AR)和拓撲異構酶Ⅱβ至TMPRSS2和ERG基因的調控區域內的斷點區域,從而誘導DNA雙鏈斷裂和基因融合;因此,這種融合被認為是由長期暴露于雄激素、AR活性增強和阻止DNA雙鏈斷裂的蛋白質PIWIL1的抑制造成的[9]。此外,TMPRSS2-ERG基因融合還受ER依賴性信號調節:在高級前列腺上皮內瘤變中,ERα上調并介導雌二醇的致癌作用,在去勢抵抗性前列腺癌中,ERβ作為腫瘤抑制因子發生部分缺失,從而促進TMPRSS2-ERG基因融合[10]。最新研究發現,BET溴結構域蛋白家族成員中的溴結構域蛋白4(Bromodomain containing 4,BRD4)參與修復DNA雙鏈斷裂和非同源末端連接途徑,促進TMPRSS2-ERG 融合[11]。
然而TMPRSS2-ERG融合基因作用機制尚不清楚。TMPRSS2的啟動子含有雄激素敏感元件,雄激素存在時,該融合基因驅動ERG過表達,調控一系列的靶基因和信號通路,從而抑制前列腺細胞分化和增加PCa細胞侵襲性,促進PCa的發生與發展。
幾乎在所有PCa中,AR都高表達。一方面,AR驅動TMPRSS2-ERG基因融合;另一方面,該融合基因反之破壞AR信號傳導,從而導致細胞處于去分化狀態和惡性轉化。此外,ERG還解離多梳抑制復合物2,生成多梳抑制復合物2亞基EZH2,上調組蛋白去乙酰化酶和致癌轉錄因子c-Myc,來抑制細胞分化,促進PCa的惡性轉化[12-13]。
ERG過表達可抑制15-羥基前列腺素脫氫酶(15-hydroxyprostaglandin dehydrogenase,HPGD),從而阻斷前列腺素E2(prostaglandin E2,PGE2)分解代謝,PGE2積聚還導致尿激酶型纖溶酶原激活物(urokinase-type plasminogen activator,uPA)活化和細胞生長,ERG還直接與uPA的啟動子結合,上調PGE2受體4(E prostanoid receptor type 4,EP4);ERG可與 SPOP(Speckle-type POZ protein)同時發生突變,促進PCa侵襲和轉移;ERG過表達上調基質金屬蛋白酶-9(matrix metalloproteinase-9,MMP-9)和PLXNB1(plexin B1)的表達[14-15];ERG 過表達激活Notch信號,從而激活Hes(Hairy/Enhancer of split)和Hey-1(hairy/enhancer-of-split related with YRPW motif protein 1),促進PCa細胞增殖。Notch信號還可上調上皮-間質轉化(epithelial-to-mesenchymal transition,EMT)轉錄因子;ERG過表達時有磷酸酶和張力蛋白同系物(phosphatase and tensin homologue,PTEN)失活或缺失,隨后通過PTEN/AKT/PIK3/mTOR途徑加速PCa的發展[16];ERG過表達導致EZH2(enhancer of Zeste Homolog 2)生成,EZH2催化組蛋白H3K9和H3K27三甲基化,招募DNA甲基轉移酶(DNA methyltransferase,DNMT);ERG過表達還上調組蛋白去乙酰化酶(histone deacetylases,HDACs)表達和抑制組蛋白乙酰轉移酶(Histone acetyltransferases,HATs)。ERG還可以結合其自身啟動子內的ETS基序,即反式激活自身啟動子,形成正反饋循環,與PCa細胞的侵襲性增加有關,見圖2。

Fig.2 The role of TMPRSS2-ERG fusion gene in the pathogenesis of prostate cancer圖2 TMPRSS2-ERG融合基因在PCa發病機制中的作用
3.1.2 TMPRSS2-ERG融合基因在PCa中的臨床研究進展 在診斷、監測進展風險和預后方面,TMPRSS2-ERG融合基因與高侵襲性和預后不良的PCa相關,免疫組織化學染色法檢測和ERG抗體可以有效診斷PCa及其轉移灶[17]。TMPRSS2-ERG融合基因檢測有助于優化臨床風險分層、預測主動監視期間PCa的進展風險、提高預后準確性;其也是PCa患者接受前列腺切除術治療最重要的預后因素[18]。聯合分析 TMPRSS2-ERG、前列腺抗原 3(PCA3)和高級別前列腺上皮內瘤變可提高特異度和敏感度,減少前列腺組織活檢[19-20]。
在治療方面,首先,靶向TMPRSS2-ERG融合基因及相關信號通路的靶向治療可以作為PCa治療的新方向。目前,恩雜魯胺(Enzalutamide)和阿比特龍均已被美國食品藥品監督管理局(FDA)批準廣泛應用于轉移性去勢抵抗性前列腺癌的治療,這2種藥物顯著提高總體生存率,但很快出現耐藥[21],因此需要更有效的靶向藥物。靶向抑制TMPRSS2-ERG融合基因表達可引起PCa細胞生長明顯停滯。BET抑制劑通過抑制BRD4表達,進而抑制DNA修復基因在前列腺細胞中的表達和非同源末端連接DNA修復途徑,從而減少TMPRSS2-ERG融合[11]。目前有兩項關于iBET762的臨床試驗正在進行中(ClinicalTrials.gov;NCT01587703,NCT01943851)。此外,ERG抑制肽和衍生肽類可以特異性結合和水解ERG蛋白,從而阻斷ERG蛋白與DNA和蛋白質之間的相互作用,治療前景廣闊[22]。
其次,Notch通路抑制劑可間接抑制TMPRSS2-ERG表達,延緩TMPRSS2-ERG陽性PCa的生長,并且提高對各種腫瘤常規療法的藥物敏感性。小分子γ-分泌酶抑制劑1是Notch通路抑制劑,可以抑制TMPRSS2-ERG陽性PCa細胞的生長,并增加其對雄激素生物合成抑制劑和AR抑制劑的敏感性[15],但其作用未完全確定[23]。另外,2種HDAC抑制劑,抗癲癇藥物丙戊酸/2-丙基戊酸(Valproic acid/2-propylpentanoic acid,VPA)和曲古抑菌素-A,可結合HDAC催化位點,從而抑制Ⅰ、Ⅱ類HDAC和AR的活性,并且影響p53的乙酰化狀態,上調p21/WAF1/CIP1,抑制TMPRSS2-ERG的表達,從而抑制PCa的發生與發展。Notch和AR抑制劑的組合療法、同時靶向EZH2和組蛋白去乙酰化酶的一些聯合治療均有治療前景[15]。
3.2 ERG在EWS中的表達及臨床研究進展 EWS是繼骨肉瘤后第二大原發惡性骨癌,治療主要依靠放療、化療和分子靶向治療[24]。85%~90%的EWS患者中存在一種特異性的t(11;22)易位,最常見的融合基因包括 EWS-FLI1[t(11;22)(q24;q12)]、EWSBR1-ERG[t(21;22)(q22;q12)]等,還有罕見的FUS-ERG融合陽性EWS。鑒于EWS細胞形態學具有非特異性,這種基因融合可以改進EWS的鑒別、分類、診斷和分子靶向治療[2]。
3.2.1 EWS中的EWSBR1-ERG和FUS-ERG融合基因及作用機制 RNA結合TET家族的N′端轉錄激活結構域可以和ETS家族的C′端DNA結合結構域發生重排。在典型EWS中,TET家族(包括EWSR1或FUS)與ETS家族(包括FLI1、ERG、ETV1、ETV4或FEV)的基因之間發生融合。最常見的融合基因是EWS-FLI1,其次是22q12上EWSR1基因與ETS家族成員之間發生融合,如ERG、ETV1、ETV4和FEV[2]。致癌融合蛋白僅在結合RNA解旋酶A之后才能具有活性。
EWSR1融合蛋白的表達干擾內源性EWSR1功能,而內源性EWSR1在促進DNA修復因子聚集到DNA損傷部位中起關鍵作用,并且通過改變選擇性剪接來降低幾種DNA損傷應答基因的表達水平。因此,EWSR1融合蛋白導致EWS中的基因組不穩定性和高水平復制應激(replication stress,RS),從而導致內源性DNA損傷[25]。攜帶致癌基因誘導的RS的EWS細胞依賴于共濟失調-毛細血管擴張有關激酶(Ataxia telangiectasi and Rad3 related,ATR)/細胞周期檢測點激酶1(Checkpoint kinase 1,CHK1)存活,在EWS中觀察到ATR和CHEK1過度表達和活化,CHK1通過下調RS和防止ATM/半胱天冬酶-3依賴性細胞死亡,從而增加EWS細胞的增殖和生存能力[26]。其次,ERG等異常的轉錄因子反式激活其他基因,參與EWS的發生與發展。如具有DNA修復、細胞分化、增殖和腫瘤轉化功能的多聚腺苷二磷酸核糖聚合酶[poly(ADP-ribose)polymerase 1,PARP-1],其啟動子中轉錄起始位點周圍存在ETS結合位點,EWSBR1-ERG和FUS-ERG融合產物驅動PARP1過表達,進而促進轉錄激活,形成正反饋環,與EWS細胞的增殖相關。此外,EWSBR1-ERG融合蛋白激活轉錄抑制因子HDAC和其他阻遏物,來抑制視黃醇類 X 受體 α(Retinoid X receptor α,RXRα)及其靶基因的正常轉錄活性。RXRs作為一種核受體與靶基因啟動子中的特定序列結合而起轉錄因子作用,可以介導類視黃醇包括天然維甲酸及其合成衍生物的抗癌功能。
3.2.2 EWSR1-ERG和FUS-ERG融合基因在EWS中的臨床研究進展 在診斷方面,熒光原位雜交技術檢測EWSR1突變是診斷EWS的金標準,但存在假陰性。因此在具有典型形態學和(或)強CD99和ERG免疫反應的情況下,應用一些分子測試可提高復雜基因重排的診斷率,例如熒光原位雜交技術或實時逆轉錄-聚合酶鏈反應檢測[27]。
在治療方面,多種靶向藥物試驗正在進行中。PARP抑制劑奧拉帕尼可通過抑制PARP1修復EWS細胞的DNA,減少EWS細胞的分裂和侵襲,但少數ETS結構域內存在點突變時無效。一項前瞻性Ⅱ期臨床試驗(ClinicalTrials.gov;NCT01583543)中,奧拉帕尼在標準化療失敗后的晚期EWS患者中安全且耐受性良好。目前,關于奧拉帕尼和替莫唑胺、伊立替康等化療藥物聯合應用的臨床試驗正在積極進行中(ClinicalTrials.gov;NCT01858168)。此外,CHK1/ATR抑制劑作為單一藥物通過多種機制增強RS,從而導致DNA損傷積累和隨后的細胞死亡,因此可作為潛在療法[25]。再者,干擾致癌融合蛋白和RNA解旋酶A相互作用的新型生物靶向小分子,例如YK-4-279,可抑制ERG和ETV1依賴的轉錄活性,從而降低EWS細胞的運動性和侵襲性[28]。最后,VPA作為HDAC抑制劑,可以抑制異常融合蛋白EWS-ERG和EWS-FLI1在EWS細胞中的表達,從而增加EWS細胞對細胞凋亡的易感性,并且減輕EWSBR1-ERG融合蛋白對RXRα轉錄活性的抑制,恢復RXRα靶基因RARβ、CRABP2和p21的抑癌活性,因此VPA具有治療潛力,但尚未見相關臨床試驗。
3.3 ERG在白血病中的表達及臨床研究進展 研究發現,在一些白血病中也出現ERG過表達,并且ERG高表達與不同類型血液惡性腫瘤的總體生存率和無病生存率相關。此外,ERG高表達還可以作為正常細胞遺傳學的急性髓系白血病(acute myeloid leukemia,AML)、T細胞急性淋巴細胞白血病(T-acute lymphoblastic leukemia,T-ALL)和兒童急性淋巴細胞白血病(Acute lymphoblastic leukemia,ALL)等獨立的預后不良因素[29-31]。
3.3.1 白血病中的ERG基因及作用機制
3.3.1.1 T-ALL 在早期T淋巴細胞生成過程中ERG表達出現下調,但在T-ALL中ERG正常表達。正常T細胞中ERG+85 kb增強子無活性,而在TALL中該增強子高度活躍,且針對造血干細胞和胸腺T細胞祖細胞,導致ERG表達。其他T細胞癌基因SCL/TAL1,LMO2以及ETS(ERG,FLI1)等轉錄因子,與ERG+85 kb的增強子結合,介導ERG高表達。
此外,ERG陽性白血病中出現幾種癌癥相關信號通路上調,其中MAPK/ERK信號通路最為明顯。MAPK/ERK信號傳導介導ERG3 S283的高度磷酸化,因此在白血病細胞中ERG3磷酸化的絲氨酸283(phosphorylation of serine 283,pS283)高于正常HSPC,并導致與MAPK/ERK活性相關的基因激活,pS283 ERG突變蛋白在ERG+85 kb增強子處富集導致內源性ERG轉錄增加,以及促進CD34+HSPC增殖[32]。
3.3.1.2 AML 研究表明,有復雜核型和21號染色體異常的AML患者均有ERG轉錄水平的上調[33]。Salek-Ardakani等[34]認為ERG作為一種癌基因可以促進各種譜系成熟造血細胞的生長,對髓系白血病發病機制和維持至關重要。與T-ALL一樣,在AML中MAPK/ERK信號通路也發揮同樣作用,導致ERG的過表達。
ERG的靶基因包括GATA2和HEY2等轉錄因子,GATA基序在ERG結合的區域內高度富集,從而導致GATA2和HEY2特異性地在ERG陽性的髓系白血病中過表達。HEY2和GATA2參與早期造血功能,GATA2是造血干細胞和MEG發育的關鍵調節因子,高GATA2表達是兒童AML不良預后因素;而HEY2被認為是心血管發育的調節因子,參與白血病的發生與進展。ERG過表達還誘導具有高度耐藥表型的間充質樣狀態,從而使腫瘤細胞獲得生長優勢[35]。
此外,有研究認為ERG通過結合PIM1的啟動子和增強子來誘導PIM1激酶癌基因表達。PIM激酶可以磷酸化組蛋白H3、c-Myc、Mcl-1、p27、Bad和FLT3,從而增加了c-Myc驅動的轉錄水平、抑制細胞凋亡、促進細胞增殖、增加FLT3-ITD介導的耐藥性,涉及癌癥進展和對化療藥物的耐藥性,高表達ERG和PIM1的AML預后較差[36]。
3.3.1.3 急性巨核細胞白血病(acute megakaryoblastic leukemia,AMKL) ERG還和AMKL有關,已發現定位于21號染色體的唐氏綜合征關鍵區域的ERG與唐氏綜合征相關的巨核細胞白血病有關。ERG是MEG生成的正調控因子,可結合SCL+19增強子,調節造血祖細胞中SCL/TAL1的表達,而SCL1/TAL1過表達會使祖細胞朝向MEG譜系分化。已知GATA1也是調控MEG生成中重要的轉錄因子,幾乎存在于所有唐氏綜合征相關AMKL患者。ERG和GATA1可影響信號通路PI3K/AKT/mTOR。活化的AKT可以與ERG、GATA1協同促進MEG異常增殖。
3.3.1.4 白血病中的FUS-ERG融合基因 除了EWS,在一些慢性粒細胞白血病急變期、急性白血病、AML 和ALL 中,也發現染色體易位 t(16;21)(p11;q22)導致了FUS與ERG融合,破壞天然的FUS RNA結合結構域,插入ERG DNA結合結構域。FUS基因位于16p11,編碼轉錄激活因子,通過結合RNA polⅡ和一些轉錄因子參與轉錄起始,從而產生異位轉錄激活。FUS外顯子1~6,7或8以及ERG外顯子的9,10或11到C′末端參與融合基因形成。FUS-ERG不僅影響骨髓譜系,還影響淋巴譜系或更原始的造血細胞。這種白血病抵抗化療且復發率高,預后很差。此外,FUS-ERG融合蛋白具有雙重轉化活性,取決于其N′末端結構。FUS-ERG融合蛋白還干擾RNA的剪接。FUS的N′末端可以結合RXR,因此FUS-ERG融合蛋白也是視黃酸信號傳導途徑的轉錄阻遏物。
3.3.2 ERG及其融合基因在白血病中的臨床研究進展 在診斷和預后方面,由于白血病皮膚和皮膚中反應性粒細胞浸潤在病理學上難以區分,而ERG可以作為用來區分的免疫組織化學標志物來區分兩者,Xu等[37]在白血病患者皮膚活檢中檢測到ERG陽性率高達81.4%(13/16),特異度為100%。另外,在ALL中,ERG與ALL中的糖皮質激素(GCs)耐藥性有關,ERG在GCs敏感細胞中被招募到GCs受體的啟動子上,這是GCs依賴性細胞凋亡所需要的,而在耐藥細胞中未觀察到這種招募,并且ERG和激活蛋白-1可以作為ALL中GCs應答的決定因素[38]。
在治療方面,PIM抑制劑SGI-1776對FLT3和PIM激酶具有雙重抑制作用,能夠降低組蛋白H3、c-Myc、Mcl-1、p27、Bad和FLT3的磷酸化,可以抑制ERG陽性的白血病細胞生長并誘導其凋亡,抑制效果與PIM1的表達水平相關[36]。但由于SGI-1776有心臟劑量限制性毒性,其臨床試驗已撤回(ClinicalTrials.gov;NCT01239108)。用小干擾RNA敲除PIM1也可以抑制ERG陽性白血病細胞的生長。此外,RAS途徑激活是ERG驅動白血病的間接靶點,PIM抑制劑和RAS抑制劑組合可增加抑制效果[36]。但在治療過程中,ERG過表達細胞會通過與基質細胞相互作用來增加細胞存活率,還可通過抑制蛋白激酶C或Wnt信號傳導途徑介導凋亡逃避,從而導致耐藥性。
鑒于ERG影響AKT并具有協同作用,AKT變構抑制劑MK-2206可以通過抑制AKT磷酸化來抑制PI3K/Akt/mTOR通路激活,誘導白血病細胞中G1期停滯和細胞凋亡。MK-2206還通過抑制糖原合酶激酶-3β磷酸化,導致糖原合酶激酶-3β介導的蛋白酶體依賴性抗凋亡促存活蛋白髓樣細胞白血病-1(myeloid cell leukemia-1,Mcl-1;Bcl-2蛋白質家族的成員)降解,從而降低白血病細胞活力、誘導細胞凋亡。并且MK2206能夠提高AML中阿糖胞苷活性。MK-2206已在復發或難治性AML患者中進行臨床試驗(ClinicalTrials.gov;NCT01253447)。
3.4 ERG在其他腫瘤中的表達以及臨床研究進展 ERG在其他腫瘤中也有一定作用。ERG在具有軟骨分化的骨和軟組織腫瘤中穩定表達,可作為軟骨分化的標志物。ERG是血管內皮以及血管結構衍生腫瘤高度敏感性和特異性的標志。ERG在所有血管瘤和淋巴管瘤、一些高度血管化的中樞腫瘤的內皮細胞中穩定表達[3],可作為高度特異性的良惡性血管性腫瘤的新標志物[47],但尚不清楚其是否有致癌作用。此外,ERG僅在中樞神經系統腫瘤的內皮細胞中表達,可作為特異性的中樞神經系統腫瘤內皮細胞標志物。ERG在上述三類腫瘤中研究較少,有待進一步探究。
4 展望
在多種腫瘤中ERG基因被發現過表達,成為目前腫瘤研究熱點。近年來,ERG在一些腫瘤中的發現以及分子機制方面的研究逐步深入,ERG本身作為癌基因或者通過融合基因在多種腫瘤中發生過表達,從而調控下游靶基因和信號通路。盡管ERG作用機制尚未完全清楚,但給腫瘤發病機制研究提供了新思路和方向。
目前,ERG已在一些腫瘤中初步顯示出治療潛力和預后價值,成為抗腫瘤治療中有前景的靶點和預后標志物。目前已有靶向治療藥物應用于臨床,還有一些正處于臨床試驗階段。但是由于ERG在正常組織中也存在表達,如何開發特異性的腫瘤靶向藥物及其相應的療效評估有待研究。在預后方面,ERG在腫瘤中可以用于監視腫瘤進展風險和提高預后準確性,顯示出一定的預后價值,但還需更多前瞻性臨床試驗來驗證。此外,鑒于ERG基因在腫瘤血管生成方面的作用,ERG在EC中高表達提示腫瘤微血管豐富,因此靶向ERG抑制腫瘤血管生成和ERG作為潛在的預后標志物均具有研究前景。綜上所述,ERG基因在部分腫瘤的發病機制、治療、預后等方面顯現出初步的研究價值,為腫瘤的發病機制研究、精準治療和預后評估提供了新的方向。
猜你喜歡
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
數學年刊A輯(中文版)(2022年4期)2022-02-16 08:17:34
今日農業(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
無線電通信技術(2021年4期)2021-07-13 08:58:28
無線電通信技術(2021年3期)2021-06-08 03:33:48
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
無線電工程(2020年11期)2020-10-29 01:25:46
現代出版(2020年3期)2020-06-20 07:10:34
福利中國(2015年4期)2015-01-03 08:03:38
