鎂對原位晶化催化劑抗鎳性能的影響
2019-03-15 08:57:30黃蕾,張莉
石油煉制與化工 2019年3期
黃 蕾,張 莉
(1.西安石油大學化學化工學院,西安 710065;2.中國石油蘭州化工研究中心)
隨著原料油重質化、劣質化和重金屬元素鎳、釩、鐵、銅等的污染加劇,對催化裂化(FCC)催化劑提出了更高的抗重金屬及重油轉化性能要求[1]。原位晶化催化劑是FCC催化劑的一種,由于采用天然礦物經過特殊工藝制備而得,使其具有優異的重油轉化和抗重金屬能力以及廣闊的市場應用前景[2-4]。在FCC反應過程中,鎳具有較強的脫氫活性,并促使不飽和烴進行縮聚反應而生焦,使干氣中氫氣產率增加,嚴重破壞了FCC催化劑的裂化選擇性[5]。并且,脫氫后的生成物會堵塞催化劑孔道,降低催化劑的表面積,影響其裂化活性。目前,大多數具有抗鎳性能的FCC催化劑都是基于改變催化劑的分子篩結構或改變催化劑基質的物理結構的原理以研制得來[6-11]。
原位晶化催化劑作為FCC催化劑的一種,由于制備工藝的特殊性,分子篩晶粒較小,酸性中心暴露充分,基質孔結構發達,使得其在重油轉化、抗重金屬等方面優勢突出,應用效果顯著[12-18]。為了增強該類催化劑的抗重金屬性能,拓寬催化劑種類,本研究在原位晶化FCC催化劑的制備過程中,采用浸漬法引入具備抗鎳功能的鎂對其進行改性,再對其進行重金屬鎳污染,借助N2吸脫附等溫線、紅外光譜、X射線衍射等手段對鎂改性后催化劑樣品進行表征,在固定流化床裝置考察鎂對原位晶化催化劑抗鎳性能的影響。
1 實 驗
1.1 主要原料及試劑
催化劑A,原位晶化催化劑,主要含有氧化硅、氧化鋁和氧化鈉,工業品,取自中國石油蘭州石化分公司;氯化鎂,分析純,國藥集團化學試劑有限公司生產;硝酸鎳,工業級,取自中國石油蘭州石化分公司。
1.2 催化劑制備
1.2.1鎂改性催化劑的制備采用浸漬法[19]將鎂負載于催化劑A。稱取一定量催化劑A,以氯化鎂為浸漬原料,浸漬量(質量分數,以鎂計)分別為0,0.5%,1.0%、1.5%,2.0%,2.5%,于120 ℃下烘干4 h,再于600 ℃下焙燒2 h,得到鎂改性催化劑A,分別標記為A0,A1,A2,A3,A4,A5。
1.2.2鎳污染鎂改性催化劑的制備將鎂改性催化劑A進行重金屬鎳污染實驗。以5 000 μg/g的硝酸鎳溶液為污染源,分別對A0,A1,A2,A3進行污染,以催化劑恰好全部潤濕為最佳,攪拌,然后將被污染的催化劑樣品在120 ℃下干燥4 h,待干燥后,在一定溫度下焙燒2 h,得到鎳污染的鎂改性催化劑,分別標記為B0,B1,B2,B3。
1.3 分析與表征
1.3.1比表面積、孔體積和孔徑的測定采用經典N2吸附-脫附等溫線法和BET法測定催化劑的比表面積、孔體積和孔徑分布,所用儀器為Autochem 2920型程序升溫脫附儀和Micromeritics ASAP-3000型自動物理吸附儀。催化劑裝填量為0.06 g左右,經300 ℃抽真空脫氣預處理8 h后,再在液氮條件下進行吸附-脫附。
1.3.2酸性表征高嶺土型催化劑表面酸性測定所用儀器為德國布魯克公司生產的布魯克-TENSOR27型紅外光譜儀,樣品壓片后需先在高真空系統中進行脫氣,選擇吡啶作為堿性吸附探針分子,在一定蒸氣壓下進行氣-固吸附。稱取0.10 g樣品壓片,在350 ℃下抽真空2 h,然后降溫到200 ℃吸附吡啶,程序升溫至測定溫度(200 ℃、350 ℃)進行真空脫附25 min,測量并記錄紅外光譜圖。
1.3.3相對結晶度測定采用XRD衍射法測定催化劑樣品的結晶度,所用儀器為日本Rigaku公司生產的D/max-2000PC型X射線衍射儀,工作電壓40 kV,電流20 mA,Cu Kα輻射,物相掃描角度5°~50°,掃描速率10 (°)/min;結晶度掃描角度22.5°~25.0°,掃描速率1 (°)/min,結晶度(X)計算式如下:
式中:HX為未知樣品的衍射峰高;H標為標準樣品的衍射峰高;w標為標準樣品的質量分數。
1.3.4微反活性的測定采用北京惠爾三吉綠色化學科技有限公司生產的MAT型微反活性評價裝置測定催化劑的微反活性,原料油為天津大港直餾輕柴油。評定條件:催化劑經800 ℃、100%水蒸氣老化17 h,原料油通入量1.560 g,反應溫度460 ℃,反應時間70 s。液體產物用Varian 3800型氣相色譜儀分析,得到催化劑的平均裂化活性,即為微反活性。
1.3.5催化劑的選擇性采用XGL-2×3型固定流化床評價催化劑的選擇性。該裝置由進樣系統、反應系統、分離采集系統和分析系統組成。反應前將樣品水熱老化處理,溫度為800 ℃,時間為6 h。然后將其裝入固定流化床,原料油取自中國石油蘭州石化分公司3.0 Mt/a重油FCC裝置,主要性質見表1。反應條件為:反應溫度500 ℃,劑油質量比5.5。采用Agilent 6890氣相色譜儀分析氣體組分和模擬蒸餾液體產物,得到裂化產品分布。
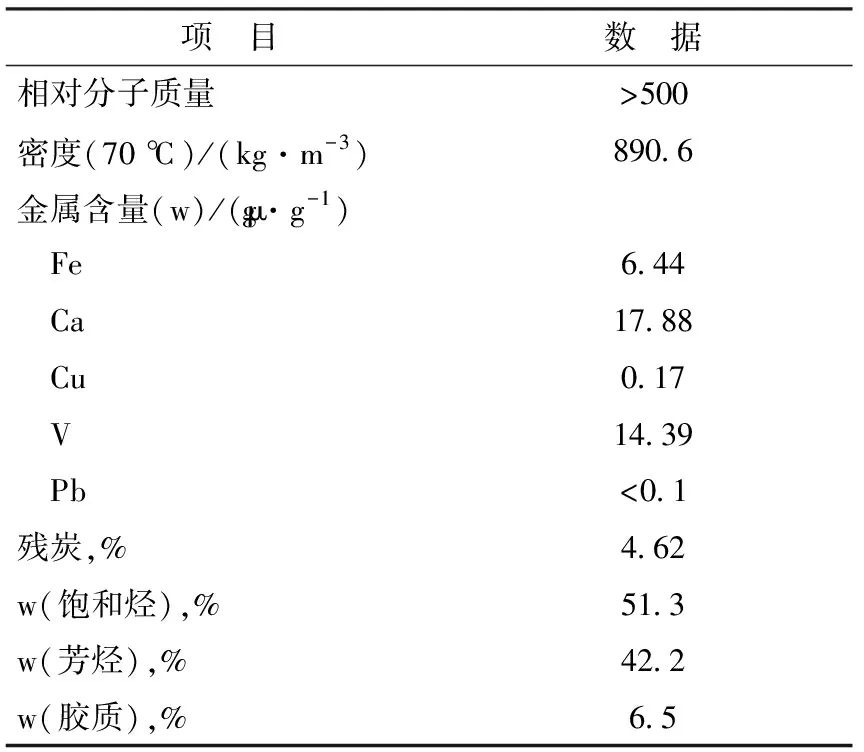
表1 原料油的主要性質
2 結果與討論
2.1 不同鎂負載量催化劑的物化性質
不同鎂負載量催化劑的物化性質見表2。從表2可以看出:①隨著鎂浸漬量的增多,催化劑樣品的比表面積先增大再降低,當鎂負載量(w)達到1.5%(A3)時,催化劑樣品的比表面積達到最大值,為369.6 m2/g;鎂負載量(w)大于2.0%時,催化劑樣品的比表面積比未負載鎂時的比表面積還小,即負載大量的鎂會使催化劑的比表面積降低;②隨著鎂浸漬量的增多,催化劑樣品的平均孔徑先減小再增大,這是由于催化劑樣品比表面積的改變引起了孔徑的變化,催化劑的比表面積越大,則孔徑越小;③催化劑上負載鎂后,相對結晶度變化并不大,當鎂負載量(w)為2.0%(A4)時,相對結晶度變化為0,即鎂負載量(w)為 2.0%時,對催化劑樣品的結晶度影響不大。
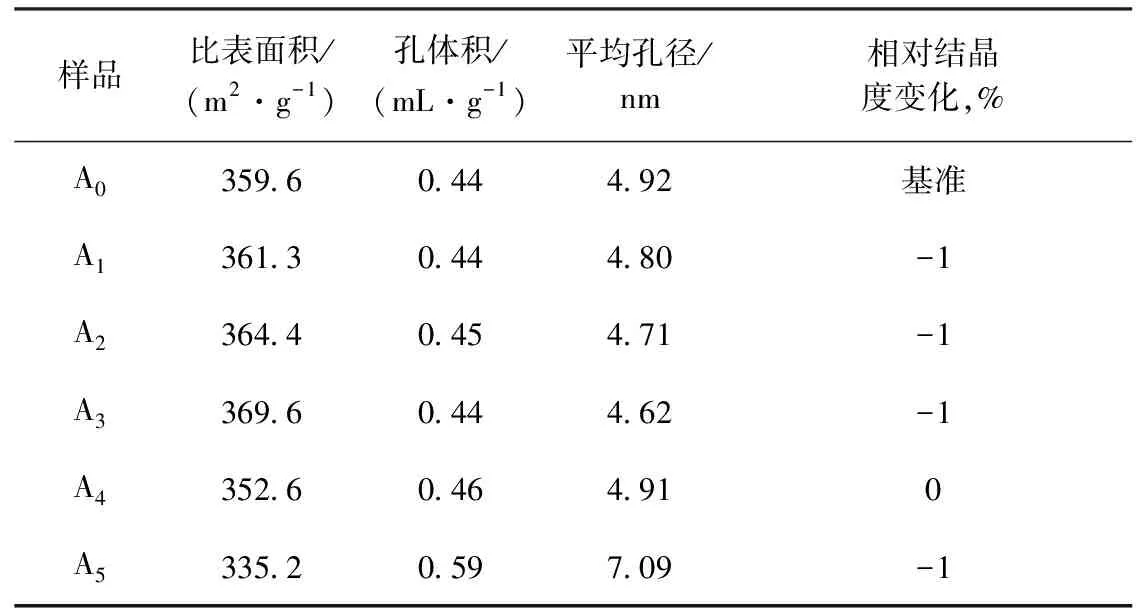
表2 不同鎂負載量催化劑的物化性質
2.2 不同鎂負載量催化劑的酸類型和酸量
不同鎂負載量催化劑樣品的酸量見表3。一般認為,200 ℃時的酸量為總酸量,350 ℃時的酸量為強酸(包含中強酸)量[20-21]。對于FCC催化劑,B酸引導正碳離子反應,強B酸中心有助于增強催化劑對重油的裂解能力,L酸主要引導自由基反應,也可引導正碳離子反應,弱L酸有助于重油大分子的裂化,因此要求催化劑有適量的弱L酸中心,并盡量減少強L酸中心[22-23]。從表3可以看出:①隨著鎂負載量的增多,催化劑樣品的B酸、L酸酸量逐漸增大,達到峰值后,逐漸降低;②隨著鎂負載量的增多,強L酸酸量增幅較小,強B酸酸量增幅較快,這可能是由于將鎂引入催化劑后,一部分負載于催化劑基質上,使得弱L酸中心有所增加;另一部分與催化劑表面的活性中心相互作用,使得強B酸中心增加;③當鎂負載量(w)為1.5%(A3)時,總酸量和強酸量均達到最大值,可能是由于鎂與催化劑表面的活性中心相互作用生成了酸性比硅鋁羥基更強的物質,使得催化劑酸性增強,說明在FCC催化劑中添加適量鎂可增強催化劑表面的酸性活性中心;④當鎂負載量(w)大于2.0%時,總酸量和強酸量減少,可能是由于當鎂負載量達到一定值后,與催化劑表面的活性中心相互作用達到飽和,催化劑表面的酸量不再增加。
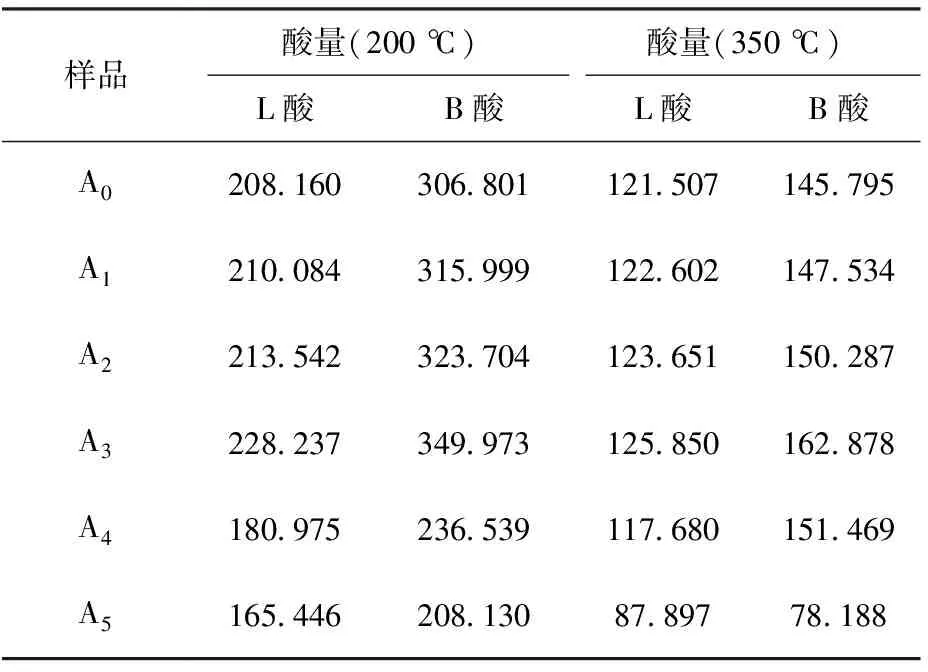
表3 不同鎂負載量催化劑樣品的酸量 μmol/g
2.3 鎳污染的鎂改性催化劑的微反活性
鎳污染的鎂改性催化劑的微反活性見表4。從表4可以看出:在未負載鎂時,微反活性最高,達到80%;隨著鎂負載量的增多,微反活性下降,當鎂負載量(w)為1.0%(B2)時,與未負載鎂時相比,催化劑的微反活性下降19百分點。說明催化劑在一定量鎳污染的情況下,當負載大量鎂時,會嚴重降低催化劑的微反活性,不利于FCC反應進行。

表4 鎳污染的鎂改性催化劑微反活性
2.4 鎳污染的鎂改性催化劑的催化反應性能
采用固定流化床對B0,B1,B2,B3樣品的催化反應性能進行評價,結果見表5。從表5可以看出:①在鎳污染條件下,催化劑上鎂的負載量為0(B0)時,轉化率最高,達到79.87%,C5+汽油收率最高,達到48.47%,柴油收率和重油收率均為最低,說明此時催化劑的裂化深度和裂化活性最強;②在鎳污染條件下,隨著催化劑上鎂負載量的增加,轉化率略有降低、C5+汽油收率變化不大、干氣和焦炭產率隨之下降,這主要是由于鎂在催化劑上的存在使其酸性發生調變的結果,也說明在催化劑上負載適量的鎂能在一定程度上抑制鎳的脫氫生焦活性,從而減輕鎳污染情況;③當催化劑上鎂負載量(w)為1.0%(B2)時,與未負載鎂的催化劑樣品(B0)相比,轉化率降低5.97百分點,C5+汽油收率降低0.01百分點,柴油收率上升3.54百分點,重油收率上升2.46百分點,干氣產率下降0.81百分點,焦炭產率下降4.36百分點,說明在相同的鎳污染條件下,催化劑上負載鎂能有效降低干氣和焦炭產率,減輕鎳對催化劑的污染,但不能提高重油轉化率和C5+汽油收率,且使得催化劑的裂化深度降低,裂化活性和選擇性減弱,從而使催化劑自身的催化反應性能變差,工業應用價值不高。
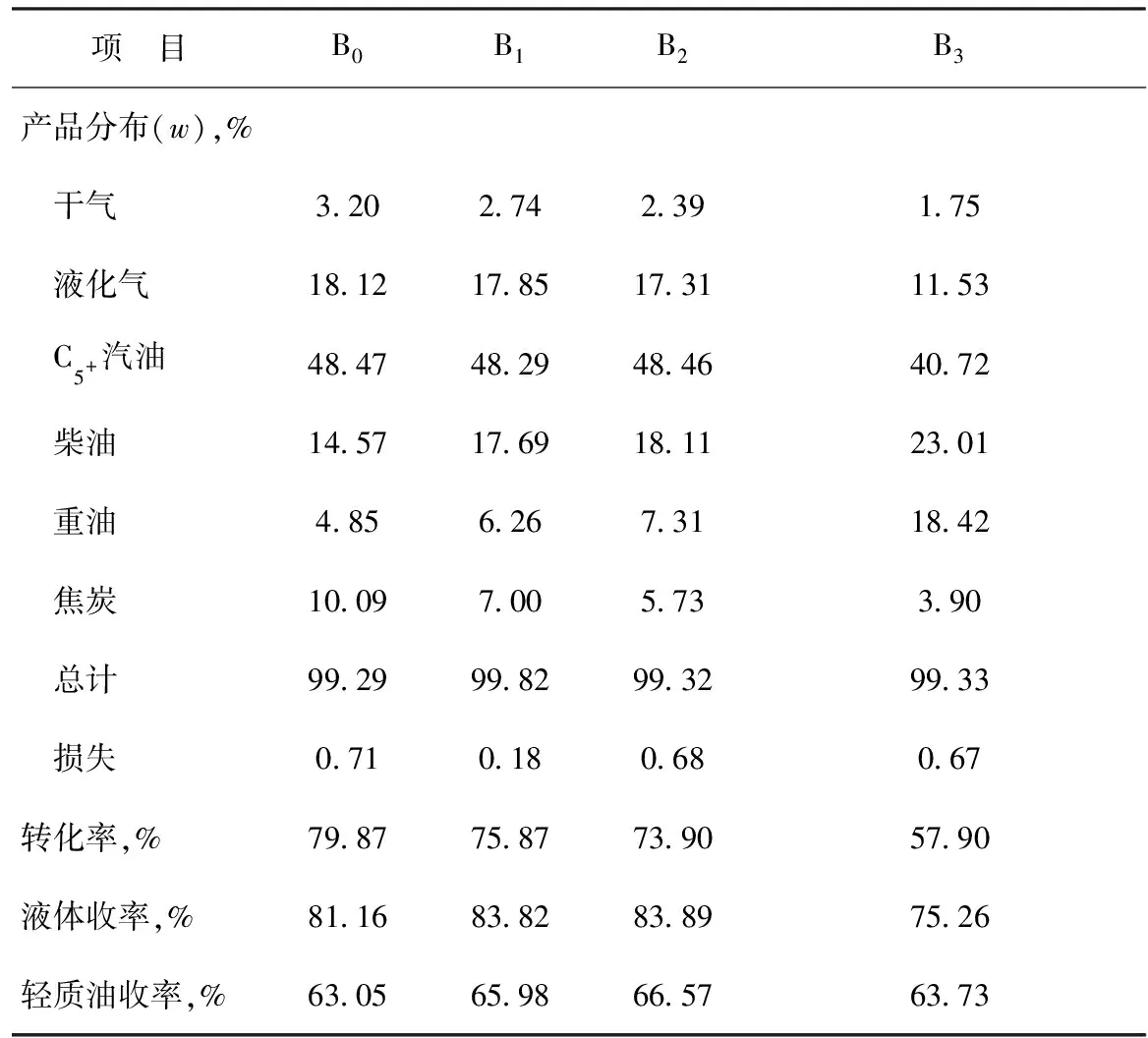
表5 鎳污染鎂改性催化劑的催化反應性能
3 結 論
(1)在原位晶化催化劑上負載適量的鎂可提高其比表面積;增加其酸性活性中心。
(2)在鎳污染條件下,隨著催化劑上鎂負載量的增多,催化劑的微反活性下降。當催化劑上鎂負載量(w)為1.0%時,與未負載鎂的樣品相比,微反活性下降19百分點,重油轉化率降低5.97百分點,C5+汽油收率降低0.01百分點,干氣產率下降0.81百分點,焦炭產率下降4.36百分點,說明在原位晶化催化劑上負載適量的鎂能夠提高催化劑的抗鎳性能。
猜你喜歡
當代陜西(2019年7期)2019-04-25 00:22:18
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
領導決策信息(2018年26期)2018-10-12 02:18:26
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
