以纖維二糖為底物利用重組大腸桿菌合成海藻糖
2019-04-12 05:34:30鄭兆娟岳泰穩歐陽嘉
食品科學 2019年6期
關鍵詞:途徑
李 新,鄭兆娟,岳泰穩,歐陽嘉*
(南京林業大學化學工程學院,江蘇 南京 210037)
木質纖維原料是地球上最豐富、最廉價的可再生資源,是制備生物燃料和生物基化學品的首選原料[1-3]。由于其結構頑固性,需要對其進行預處理和酶水解才能得到微生物可以利用的可發酵性糖[4-5]。其中,纖維二糖是纖維素酶水解纖維素時的主要產物。由于絕大多數微生物不能直接利用纖維二糖,需要外源添加β-葡萄糖苷酶進一步水解纖維二糖為葡萄糖,這進一步增加了酶制劑成本。此外,纖維二糖是纖維素酶的非競爭性抑制劑,強烈抑制纖維素酶的水解作用,極大地影響了同步糖化發酵效率[6-8]。因此,近年來,研究人員致力于開發能利用纖維二糖的微生物生產各種高附加值化學品。例如,Adachi等[9]通過將β-葡萄糖苷酶展示在谷氨酸棒狀桿菌細胞表面,使得谷氨酸棒桿菌具備纖維二糖水解能力,實現了纖維二糖至L-賴氨酸的發酵生產。Mu?oz-Gutiérrez等[10]使用屬于V型分泌系統的自動展示系統AIDA-I,在產乙醇的大腸桿菌菌株MS04的外膜上展示來自嗜熱放線菌的β-葡萄糖苷酶,以纖維二糖為底物,乙醇產率達到了理論最大值的81%。Soma等[11]通過將異丙醇合成途徑導入到大腸桿菌,并在大腸桿菌外膜上展示β-葡萄糖苷酶,證實了利用大腸桿菌發酵纖維二糖生產異丙醇的可行性。
海藻糖是由兩個葡萄糖分子以α,α-1,1-糖苷鍵結合的非還原性雙糖,廣泛存在于細菌、真菌和動植物體內。海藻糖不僅可以作為生物體的結構成分和能量物質,而且在氧化或干燥等極端條件下對生物體和生物大分子具有非特異性保護作用。海藻糖在食品、化妝品、農業和制藥工業中有著廣泛的應用前景[12-13]。例如,海藻糖可用于疫苗的穩定和器官的保存[14-15]。Martinetti等[16]建立了一種使用海藻糖冷凍保存純的外周血干細胞的方法,與正常冷凍保存手段相比,該方法提高了解凍后的細胞存活率。Vílchez等[17]篩選了一系列微生物作為耐干燥保護劑以保護植物免受干旱的影響,研究發現這些微生物產生的海藻糖是保護植物免受干旱的重要物質。隨著市場對海藻糖的需求不斷增大,研究人員也在不斷開發合成海藻糖的新方法。生物體內研究最多的海藻糖合成途徑主要有三條:一是OtsAB途徑,該途徑以葡萄糖為底物,通過6-磷酸海藻糖合成酶和6-磷酸海藻糖磷酸酯酶催化形成海藻糖[18-19];二是TreYZ途徑,該途徑以麥芽糊精為底物,通過麥芽寡糖基海藻糖合成酶和麥芽寡糖基海藻糖水解酶催化形成海藻糖[20];三是TreS途徑,該途徑以麥芽糖為底物,通過海藻糖合酶催化形成海藻糖[21]。
假單胞菌是一種條件致病菌,其可以利用多種不同的策略抵抗干旱等惡劣環境,其中的一種策略就是在細胞內合成海藻糖[22-23]。Zheng Zhaojuan等[21]克隆了施氏假單胞菌(Pseudomonas stutzeri)A1501來源的treS基因,并在大腸桿菌(Escherichia coli)中重組表達,全細胞催化實驗表明該重組菌株具有高效轉化麥芽糖為海藻糖的能力。基因組分析表明,P.stutzeriA1501基因組上還具備海藻糖合成的OtsAB途徑和TreYZ途徑。本研究克隆了來自P. stutzeriA1501的otsAB基因,外源導入E. coliBL21(DE3)構建合成海藻糖的OtsAB途徑。并通過過表達來自大腸桿菌的尿苷二磷酸(uridine diphosphate,UDP)-葡萄糖焦磷酸化酶基因galU來強化合成海藻糖的OtsAB途徑。在此基礎上,引入來自1 株天然纖維素分解細菌Saccharophagus degradans的纖維二糖磷酸化酶基因cepA,使大腸桿菌具備纖維二糖代謝能力。重組大腸桿菌以纖維二糖為底物合成海藻糖的代謝途徑如圖1所示。最后,本研究探討了該重組大腸桿菌以纖維二糖為底物全細胞催化合成海藻糖的可行性,為纖維二糖來源精細化學品的生產提供了新的思路。

圖1 重組大腸桿菌纖維二糖合成海藻糖的途徑Fig. 1 Biosynthesis pathway of trehalose from cellobiose in recombinant E. coli
1 材料與方法
1.1 材料與試劑
1.1.1 菌株及質粒

表1 本研究使用的菌株和質粒Table 1 Strains and plasmids used in this study
本研究所用菌株和質粒見表1,聚合酶鏈式反應(polymerase chain reaction,PCR)擴增所用引物序列見表2。其中纖維二糖磷酸化酶基因cepA(GenBank NC_007912,范圍:1710240~1712675)按照大腸桿菌密碼子偏好性優化后,由上海捷瑞生物工程有限公司合成。

表2 本研究使用的PCR擴增引物Table 2 Sequences of PCR primers used in this study
1.1.2 酶與試劑
PfuDNA聚合酶、限制性內切酶、T4 DNA連接酶TaKaRa(大連)公司;酵母提取物、胰蛋白胨 德國Sigma公司;異丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)、氨芐青霉素鈉、硫酸卡那霉素、井岡霉素 生工生物工程(上海)股份有限公司;基因組提取試劑盒 北京全式金生物技術有限公司;質粒小量快速提取試劑盒和瓊脂糖凝膠DNA回收試劑盒 上海捷瑞生物工程公司;其余試劑均為國產分析純;引物由上海捷瑞生物工程公司合成。
1.2 儀器與設備
MasterCycler Gradient 5331梯度PCR儀、5415R高速冷凍離心機 德國Eppebdorf公司;DYY-8B核酸電泳儀、GIS system凝膠成像系統 上海天能科技有限公司;752S型紫外-可見分光光度計 上海棱光技術有限公司;ICS·3000型離子色譜儀 美國Dionex公司;MSC1.2無菌操作臺 美國Thermo公司。
1.3 方法
1.3.1 分子克隆
DNA酶切、連接、感受態細胞制備和轉化、基因組提取等基本基因操作技術參照文獻[24]及供應商提供的操作手冊進行。
質粒pETDuet-otsA-otsB構建:參照GenBank數據庫中已公布的P. stutzeriA1501的otsA基因序列(GenBank NC_009434,范圍:3495671~3497086)和otsB基因序列(GenBank NC_009434,范圍:3497103~3497879),使用引物otsA-F、otsA-R擴增otsA基因插入質粒pETDuet-1的BamHI、HindIII位點,獲得重組質粒pETDuet-otsA。然后再使用引物otsB-F、otsB-R擴增otsB基因插入質粒pETDuet-otsA的NdeI、XhoI位點,獲得重組質粒pETDuetotsA-otsB,測序驗證。
質粒pRSFDuet-galU-cepA的構建:參照GenBank數據庫中已公布的E. coliBL21的galU基因序列(GenBank NC_012892,范圍:1279243~1280151),使用引物galU-F、galU-R擴增galU基因插入質粒pRSFDuet-1的BamHI、HindIII位點,獲得重組質粒pRSFDuet-galU。然后參照GenBank數據庫中已公布的S. degradans2-40的cepA基因序列(GenBank NC_007912,范圍:1710240~1712675),對cepA基因序列進行密碼子優化,送去上海捷瑞生物工程有限公司合成。使用引物cepA-F、cepA-R擴增合成的cepA基因插入質粒pRSFDuetgalU的BglII、XhoI位點,獲得重組質粒pRSFDuet-galU-cepA,測序驗證。
1.3.2 培養基與培養條件
LB培養基:胰蛋白胨10.0 g/L,酵母粉5.0 g/L,NaCl 10.0 g/L,pH 7.0。
磷酸鹽緩沖液(phosphate buffer saline,PBS):Na2HPO4·12 H2O 19.100 8 g/L,KH2PO41.815 6 g/L。
大腸桿菌培養時根據需要加入終質量濃度為100 mg/L的氨芐青霉素鈉或40 mg/L的硫酸卡那霉素。
1.3.3 重組大腸桿菌全細胞催化合成海藻糖
1.3.3.1 菌種活化
將保存于-80 ℃的菌種接入裝有5 mL LB的搖管中,接種量1%,并根據需要加入終質量濃度100 mg/L氨芐青霉素鈉,40 mg/L硫酸卡那霉素,放置于37 ℃、200 r/min條件下過夜培養。
1.3.3.2 擴培和誘導
將活化好的菌種轉入裝有1/5裝液量的500 mL的三角搖瓶中,接種量1%,并根據需要加入終質量濃度100 mg/L氨芐青霉素鈉、40 mg/L硫酸卡那霉素,放置于37 ℃、200 r/min條件下培養,OD600nm達到0.6~0.8時加入終濃度1 mmol/L的IPTG,轉移到30 ℃、200 r/min條件下繼續培養8 h。8 000 r/min離心收集誘導好的菌體,用PBS洗滌菌體2 遍,重懸于PBS中備用。
1.3.3.3 全細胞催化
全細胞催化在150 mL的錐形瓶中進行,裝有10 mL催化反應混合液,混合液中的菌體OD600nm為20,葡萄糖或纖維二糖質量濃度為20 g/L,并根據需要加入終濃度0.05 mmol/L井岡霉素。反應在30 ℃、200 r/min的條件下進行,定時取樣測定底物和產物濃度。
1.3.4 指標檢測
菌體密度測定采用比濁法。將培養液用水稀釋適當的倍數,使OD600nm值在0.6~1.0之間,進行濁度測定。測定條件:采用紫外-可見分光光度計,波長600 nm。
葡萄糖、纖維二糖、海藻糖質量濃度測用采用離子色譜法[25]。取1 mL反應混合液,室溫下12 000 r/min離心2 min,取上清液,用孔徑為0.22 μm的無菌濾膜過濾。分析條件:采用離子色譜儀,分析柱為CarboPacTM10(250 mm×2 mm);檢測方式:四電位脈沖安培檢測。以超純水、200 mmol/L NaOH(A)和500 mmol/L NaAc(B)為淋洗液進行洗脫。0~7.00 min,采用75%超純水和25% A相混合液淋洗,7.01~10 min變為100% A相,然后A相線性減少,B相線性增加,至15.00 min時,變為90% A相和10% B相混合液;15.01~30 min恢復為75%超純水和25% A相沖洗系統。進樣量為10 μL,流速為0.3 mL/min,柱溫為30 ℃。與標準曲線對照計算各物質的含量。數據測定后取平均值,本研究中誤差線代表3 個平行實驗的標準偏差。
2 結果與分析
2.1 otsA、otsB、galU和cepA基因克隆和重組質粒的構建

圖2 otsA、otsB、galU和cepA基因PCR擴增產物電泳圖Fig. 2 Electrophoretograms of PCR-amplified otsA, otsB, galU and cepA

圖3 重組表達質粒pETDuet-otsA-otsB、 pRSFDuet-galU和pRSFDuet-galU-cepA雙酶切驗證Fig. 3 Double restriction enzyme digestion analysis of recombinant plasmid pETDuet-otsA-otsB, pRSFDuet-galU and pRSFDuet-galU-cepA
提取P. stutzeriA1501的基因組,以其為模板分別PCR擴增得到otsA和otsB基因,按照1.3.1節所述實驗方法將其連接至pETDuet-1表達載體,獲得重組表達質粒pETDuetotsA-otsB并送至安徽通用生物有限公司測序。提取E. coliBL21的基因組,以其為模板PCR擴增得到galU基因,按照1.3.1節所述實驗方法將其連接至pRSFDuet-1表達載體,獲得重組表達質粒pRSFDuet-galU并送至安徽通用生物有限公司測序。cepA基因由上海捷瑞生物工程有限公司按照大腸桿菌密碼子優化后序列合成,之后以其合成基因為模板進行PCR擴增,按照1.3.1節所述實驗方法將其連接至pRSFDuet-galU,獲得重組表達質粒pRSFDuetgalU-cepA并送至安徽通用生物有限公司測序。otsA、otsB、galU和cepA基因的PCR擴增如圖2所示。瓊脂糖凝膠電泳和測序結果均表明,otsA基因長度為1 416 bp,otsB基因長度為777 bp,galU基因長度為909 bp,cepA基因長度為2 436 bp,其中otsA、otsB和galU基因序列與NCBI公布序列大小相符,序列一致性為100%。cepA基因與密碼子優化后的序列完全一致。重組表達質粒pETDuet-otsA-otsB、pRSFDuet-galU和pRSFDuet-galU-cepA雙酶切驗證如圖3所示,otsA、otsB、galU和cepA基因均已成功連接。
2.2 以葡萄糖為底物全細胞催化合成海藻糖
雖然有報道表明大腸桿菌具備海藻糖合成的OtsAB途徑[26],但可能由于該途徑太弱,無法檢測到海藻糖產量。因此本研究克隆了來自P. stutzeriA1501的otsAB基因,在E. coliBL21(DE3)內重構了一條合成海藻糖的途徑。OtsAB途徑合成海藻糖的前體物質是葡萄糖-6-磷酸和UDP-葡萄糖(圖1),由于菌體細胞自身合成的UDP-葡萄糖在細胞內積累水平較低,本實驗通過過表達大腸桿菌內參與UDP-葡萄糖合成的關鍵酶UDP-葡萄糖焦磷酸化酶來提高重組菌株細胞內的UDP-葡萄糖水平。以葡萄糖為底物全細胞催化合成海藻糖,比較出發菌株E. coliBL21(DE3)和重組菌株E. coliBL21(A),E. coliBL21(C)利用葡萄糖合成海藻糖的能力,結果如圖4所示。

圖4 對照菌與重組菌全細胞催化合成海藻糖的過程分析Fig. 4 Time course of trehalose synthesis catalyzed by whole control and recombinant cells
從圖4可以看出,出發菌株E. coli BL21(DE3)不能將葡萄糖轉化為海藻糖。重組菌株E. coli BL21(A)可以將葡萄糖轉化為海藻糖,海藻糖產量在8 h達到最高值0.25 g/L左右,這說明外源otsA/otsB基因可在大腸桿菌中成功表達并發揮功能。與E. coli BL21(A)相比,重組菌株E. coli BL21(C)轉化葡萄糖為海藻糖的能力大幅提高,海藻糖的產量在24 h達到0.75 g/L左右。過表達來自大腸桿菌的UDP-葡萄糖焦磷酸化酶后,雖然葡萄糖利用速率減慢,但是增加了海藻糖合成前體UDP-葡萄糖的含量,海藻糖的產量提高了3 倍左右。
2.3 井岡霉素對海藻糖合成的影響
大腸桿菌細胞內含有降解海藻糖的酶,即海藻糖酶,可以分解海藻糖為兩分子葡萄糖[27],從圖4可以看出,海藻糖產量在達到一定程度后開始下降,這也進一步驗證了海藻糖酶的存在。據報道[28],井岡霉素可以降低海藻糖酶的活性,抑制海藻糖在菌體內的降解。為進一步提高海藻糖的產量,在添加井岡霉素的條件下,以葡萄糖為底物全細胞催化合成海藻糖,比較重組菌株E. coli BL21(A),E. coli BL21(C)合成海藻糖的能力,結果如圖5所示。

圖5 井岡霉素對重組菌全細胞催化合成海藻糖的影響Fig. 5 Effect of validamycin A on the synthesis of trehalose
由圖5可以看出,隨著底物葡萄糖的不斷消耗,海藻糖也在不斷積累,E. coli BL21(A)轉化葡萄糖生成海藻糖的產量在36 h達到0.68 g/L左右,E. coli BL21(C)轉化葡萄糖生成海藻糖的產量在36 h達到1.1 g/L左右。同時,由圖4和圖5對比可以看出,加入井岡霉素后,海藻糖的產量得到明顯提高,并且維持在一定的水平不再下降。
2.4 以纖維二糖為底物全細胞催化合成海藻糖
據Sekar等[29]報道,S. degradans 2-40是一株天然纖維素分解細菌,可依靠cepA基因將纖維二糖磷酸解為葡萄糖和葡萄糖-1-磷酸。因此,在上述實驗的基礎上,在重組菌株E. coli BL21(C)胞內進一步過表達cepA基因,以期實現纖維二糖至海藻糖的合成通路。在添加井岡霉素的條件下,以纖維二糖為底物全細胞催化合成海藻糖,比較重組菌株E. coli BL21(C)和重組菌株E. coli BL21(D)利用纖維二糖合成海藻糖的能力,結果如圖6所示。
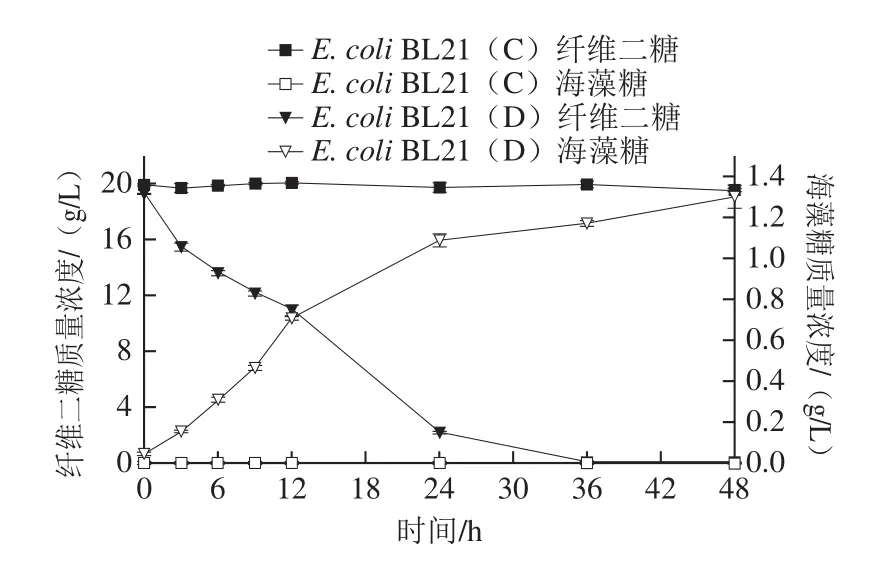
圖6 以纖維二糖為底物合成海藻糖的過程曲線Fig. 6 Time course of trehalose synthesis from cellobiose
由圖6可以看出,重組菌株E. coli BL21(C)不能將纖維二糖轉化為海藻糖,重組菌株E. coli BL21(D)可以將纖維二糖轉化為海藻糖,海藻糖的產量在48 h達到最高值1.3 g/L左右,這是以纖維二糖為底物合成海藻糖的首次報道。但是,由于大腸桿菌糖酵解途徑的存在,使得大量葡萄糖-6-磷酸進入糖酵解途徑被消耗,這導致海藻糖合成的前體物質大為減少(圖1),因此下一步應該采取合適的代謝工程策略,在不影響菌株存活的前提下,增加纖維二糖至海藻糖的代謝流量。
目前,海藻糖的生產方法主要是酶合成法和微生物發酵法。依賴于麥芽寡糖基海藻糖合成酶和麥芽寡糖基海藻糖水解酶的酶法合成途徑是最早建立的規模化海藻糖生產方法,但該方法生產工藝復雜、生產成本高,無法滿足市場對海藻糖的大量需求[30]。微生物發酵法主要是以篩選的野生型菌株、過表達6-磷酸海藻糖合成酶和6-磷酸海藻糖磷酸酯酶或者過表達海藻糖合酶的基因工程菌株作為生物催化劑,以生長體系或者全細胞催化體系實現海藻糖的合成[19]。其中海藻糖合酶的優點是只需要一步反應即可以實現麥芽糖至海藻糖的轉化,缺點是海藻糖合酶具有一定的麥芽糖水解活性,該反應常伴有副產物葡萄糖的產生[21]。依賴于6-磷酸海藻糖合成酶和6-磷酸海藻糖磷酸酯酶的OtsAB途徑在自然界分布最為廣泛,是研究最多的一種胞內合成海藻糖的方式。微生物利用OtsAB途徑合成海藻糖的底物為葡萄糖,近年來,研究者開發了多種不同的底物通過OtsAB途徑實現了海藻糖的合成。例如,Li He等[28]通過在E. coli MG1655過表達來自E. coli DH5α的OtsAB途徑,以甘油為底物實現了海藻糖的生產。Habe等[31]通過利用篩選的Burkholderia stabilis LA20W為出發菌株,以乙酰丙酸為底物合成了海藻糖。Kar等[32]利用專性嗜鹽菌Actinopolyspora halophile MTCC 263為出發菌株,以酸乳清為底物合成了海藻糖。本研究構建了以纖維二糖為底物合成海藻糖的途徑,這為以木質纖維原料為底物合成海藻糖提供了研究基礎。但是,和其他利用OtsAB途徑合成海藻糖的菌株一樣,海藻糖的產量不高,后續工作中需要結合代謝工程手段敲除支路途徑以提高海藻糖的產量。
3 結 論
本研究克隆了來自P. stutzeri A1501的otsA/otsB基因,成功構建用于海藻糖生產的重組大腸桿菌。通過增加海藻糖合成前體物質的含量和抑制海藻糖的降解,進一步提高海藻糖的產量。UDP-葡萄糖是合成海藻糖的前體物質之一,它是提高海藻糖產量的限制因素之一,本實驗通過提高胞內UDP-葡萄糖水平,使海藻糖的產量提高了3 倍。同時,菌體內還有降解海藻糖的酶系,通過添加一定濃度的井岡霉素抑制海藻糖酶的活性,進一步使提高海藻糖產量。最后,為實現菌株對纖維二糖的利用,在前期改造的菌株中進一步過表達了纖維二糖磷酸化酶基因,使重組大腸桿菌具有利用纖維二糖產海藻糖的能力,最高產量為1.3 g/L。
猜你喜歡
語數外學習·高中版中旬(2023年2期)2023-05-10 13:26:53
語數外學習·高中版中旬(2022年5期)2022-07-13 20:47:51
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
中學生百科·大語文(2017年10期)2017-11-04 06:56:38
中國衛生(2016年3期)2016-11-12 13:23:26
公民與法治(2016年22期)2016-05-17 04:20:13
中國衛生(2014年12期)2014-11-12 13:12:52
癌變·畸變·突變(2014年6期)2014-02-27 06:15:03
