正交試驗優選痔瘺寶洗劑的提取工藝研究※
2019-05-14 06:42:26謝鍇標吳美珠邱新華陳震堯
中國藥物經濟學 2019年4期
謝鍇標 吳美珠 邱新華* 陳震堯 劉 燕
痔瘺是臨床上多發病、常見病,其主要臨床表現為疼痛、便血、脫出、局部分泌物增多和排便困難等,中醫治療以坐浴為主[1]。痔瘺寶洗劑是肇慶市中醫院肛腸科專家經過多年治療肛腸疾病總結篩選出的經驗方,經劑型改革研制而成,具有消腫、止痛、止血、祛濕功效,用于治療肛痔炎癥、肛裂潰爛出血、肛門濕疹搔癢等療效顯著。痔瘺寶洗劑由大黃、兩面針、苦參、地榆、白芷、芒硝六味中藥煎煮濃縮而成。方中大黃、兩面針活血消腫止痛為君藥,苦參清熱祛濕止癢、地榆涼血止血收斂生肌為臣藥,白芷消腫散結、芒硝清熱解毒為佐藥。《本草綱目》中記載大黃能“諸火瘡”,《本草新編》中 記載大黃“破癥結,散堅聚”,《日華子本草》中記載大黃能“切瘡疔癰毒”。現代藥理學研究表明,大黃具有殺滅病原微生物、抗炎、抗腫瘤作用[2]。其抗菌機制為大黃素可破壞細菌細胞膜通透性,抑制細菌內的蛋白質合成[3]。因此,本試驗以大黃素含量和干浸膏得率為評價指標,采用正交試驗方法優化提取工藝,以期為該制劑的規模生產提供參考。
1 儀器與試劑
1.1 儀器
Agilent 1260-LC 型液相色譜儀、JA2003N 型電子天平(上海精密科學儀器有限公司);RS-FS1401 型多功能粉碎機(合肥榮事達小家電有限公司);KQ5200DE 型數控超聲波清洗儀(東莞市科橋超聲波設備有限公司);FA1004N 電子天平(上海精密科學儀器有限公司);1601 型電陶爐(廣東艾詩凱奇智能科技有限公司);JC101 型電熱鼓風干燥箱(南通嘉程儀器有限公司);420-A 型電熱恒溫水浴鍋(姜堰市新康醫療器械有限公司);ZF1-I 型多功能紫外分析儀(上海和勤分析儀器有限公司)。
1.2 試劑與試藥
大黃素(中國食品藥品檢定研究院,批號:110756-201512);芒硝(廣州至信中藥飲片有限公司,批號:180701);大黃(肇慶得隆中藥飲片廠,批號16118014);苦參(廣東新興永祥中藥飲片廠,批號:180201);地榆(廣州至信中藥飲片有限公司,批號:180701);兩面針(廣東新興永祥中藥飲片廠,批號:180801);白芷(肇慶得隆中藥飲片廠,批號:DL150170323);痔瘺寶洗劑(肇慶市中醫院,批號:180329、180716、181015);甲醇LC-MS(天津市四友精細化學品有限公司,批號:170714);其他試劑為分析純,水為純化水。
2 方法與結果
2.1 提取工藝
根據處方特點和臨床需要,該制劑用途為稀釋后外洗或坐浴,故依據傳統工藝全方用水煎煮后濃縮處理。為達到原料藥的均一性,試驗用的所有原料藥打碎為過10 目篩的粗粒,混合均勻后再按試驗量分為若干份。
2.2 設計正交試驗
根據藥材提取工藝中的加水倍數(A)、提取時間(B)、提取次數(C)等因素考察結果[4](因素水平見表1),以大黃素的含量和干浸膏得率為指標,進行正交試驗。評價指標的綜合評分:以提取液中大黃素含量和干浸膏得率為標價指標,采用公式Z[5]=(60/大黃素含量max)×大黃素含量+(40/干浸膏得率max)×干浸膏得率)進行計算,使用統計軟件SPSS 22.0 對數據進行分析。
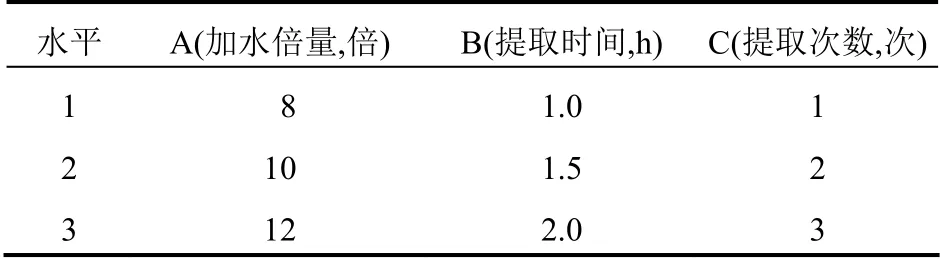
表1 因素水平
2.3 測定干膏得率
用移液管精密量取供試品25 ml 置蒸發皿,在水浴鍋中水浴蒸干后置105 ℃烘箱內干燥至恒重。得膏率(%)=干膏重量/原藥材重量×100%。
2.4 大黃素的測定
2.4.1 色譜條件以十八烷基硅烷鍵合硅膠為填充劑;以甲醇為流動相A,以0.1%磷酸溶液為流動相B[6],按表2中規定進行梯度洗脫;檢測波長為254 nm[7],柱溫30 ℃,理論板數按大黃素計算應不低于5350。見表2。

表2 痔瘺寶洗劑測定大黃素的色譜條件
2.4.2 對照品溶液制備精密稱量大黃素對照品,加入甲醇制備成大黃素儲備液,濃度為720 μg/ml。準確量取儲備液1 ml 置于25 ml 容量瓶中,加入流動相溶液定容至刻度線,充分混合均勻后,用0.45 μm微孔濾膜過濾,即得對照品供試液,大黃素濃度為57.6 μg/ml。
2.4.3 供試品溶液制備取本品20 ml 置三角錐瓶中,加鹽酸調節pH 值至2,水浴回流加熱30 min,立即冷卻,用乙酸乙酯振搖萃取3 次,每次20 ml,合并乙酸乙酯液,用蒸發皿蒸干,殘渣加甲醇定容至10 ml,作為供試品溶液。
2.4.4 陰性對照樣品溶液制備用缺大黃的陰性供試品[8],加鹽酸調節pH 值至2,水浴回流加熱30 min,立即冷卻,用乙酸乙酯振搖萃取3 次,每次20 ml,合并乙酸乙酯液,用蒸發皿蒸干,殘渣加甲醇定容至10 ml,作為陰性對照樣品溶液。
2.4.5 專屬性試驗分別抽取供試品、大黃素對照品、痔漏寶洗劑陰性(缺大黃)對照品5 μl 進入液相檢測,結果如圖1所示,表明在大黃素對照出峰的保留時間為15 min 左右,供試品同樣出峰,且分離度好,陰性無干擾。
2.4.6 線性范圍考察分別量取2.4.2 項下對照品儲備液[9],稀釋成14.4 μg/ml、28.8 μg/ml、43.2 μg/ml、57.6 μg/ml、72.0 μg/ml,按2.4.1 項下色譜條件進樣, 以進樣濃度(μg)作為橫坐標,峰面積作為縱坐標,進行回歸方程的計算,得到大黃素的回歸方程:y=19.688x-0.4001,r=0.9999。表明大黃素在72.0~360.0 ng 范圍內呈現出良好的線性關系。

圖1 HPLC 色譜圖(A 為對照品;B 為供試品;C 為陰性對照)
2.4.7 精密度試驗精密量取對照品5 μl,每隔0.5 小時進樣1 次,共6 次[10],結果顯示相對標準偏差(RSD)=0.75%,表明儀器精密度良好。
2.4.8 重復性試驗按2.4.3 項下方法,同法制備6 份供試品溶液[11],精密量取5 μl,分別進樣,結果顯示6 份供試品的大黃素含量的RSD=1.03%,結果表明本試驗條件的重復性較好。
2.4.9 穩定性試驗精密量取對照品5 μl,每隔2 小時進樣1 次,記錄大黃素的檢測峰面積,結果RSD= 0.91%,表明配制溶液8 h 內穩定性良好[12]。
2.5 正交試驗測定結果由表3綜合評分可知提取條件對工藝的影響[13]大小依次為C>B>A,提取的最佳條件為A2B3C3。從表4方差分析得知,提取次數(C)對工藝有顯著性影響,A 和B 對工藝的影響不顯著,考慮到大生產降低成本和縮短生產周期,選擇A2B2C3,即加水量為原藥材10 倍、提取3 次,每次1.5 h。
為確定本試驗的準確性,按上述確定提取方法[14],即加水量10 倍,提取3 次,每次1.5 h,結果測定出大黃素平均55.33 μg/ml(n=3,RSD=1.05%)。由驗證結果可知,本方案準確有效。
3 討論
以大黃素為內參[15-16],驗證一測多評大黃五種成分的方法是合理可行的,故本文選擇大黃素為檢測指標具有重要意義;該制劑為純水提物,干浸膏得率能整體反映提取的效率,具有現實意義。
2015 版《中華人民共和國藥典》中,大黃的提取方法是采用回流放冷后用乙醚萃取,筆者考慮到乙醚屬于“易制毒”試劑,購買時需要至公安局進行,購買手續繁雜,不便于以后實際工作,故使用溶劑極性排行表[17]與乙醚靠近的乙酸乙酯進行萃取。用乙酸乙酯替代乙醚的試驗方法是首先采用薄層色譜法,即以硅膠G 作固定相,用普通毛細管將乙酸乙酯和乙醚萃取樣品在同一硅膠G 薄層板上進行點樣,結果在相同的位置呈現的同等數量和顏色斑點是一致的;其次是采用高效液相分析所提取的大黃素含量,即采用自動進樣器分別吸取5 μl 乙酸乙酯和乙醚萃取的樣品進樣,結果二者的色譜峰面積基本一致,故使用乙酸乙酯替代乙醚萃取大黃素方法是可行的。
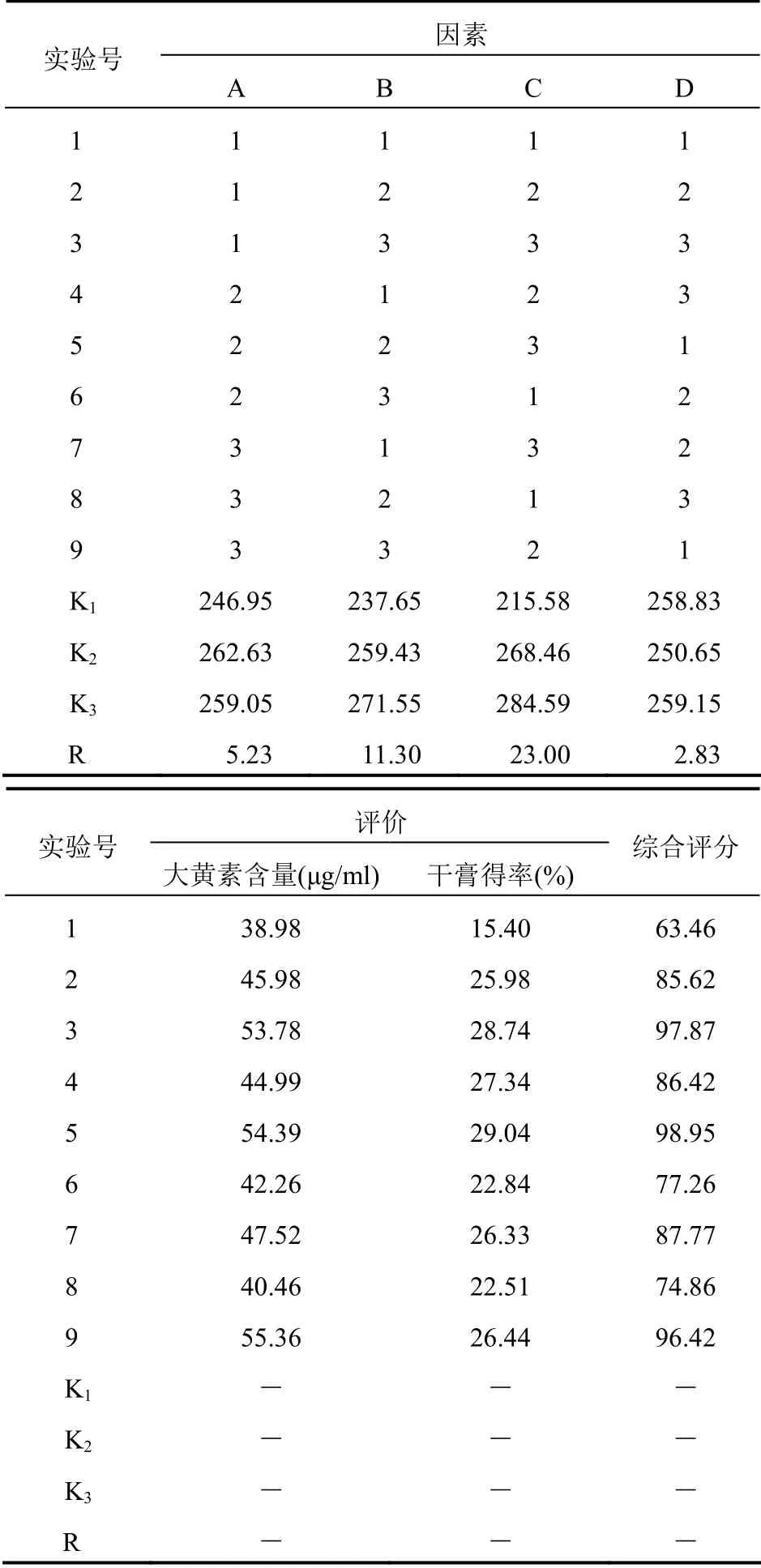
表3 正交試驗結果

表4 方差分析
在提取大黃素過程中,筆者曾使用乙酸乙酯萃取后分別上硅膠柱,分別用50%甲醇洗脫、石油醚∶乙醚8∶2 洗脫、石油醚∶乙酸乙酯5∶5 洗脫、石油醚∶乙酸乙酯3∶7 洗脫、石油醚∶乙酸乙酯1∶9 洗脫,結果石油醚∶乙醚8∶2 洗脫時供試品前面的雜質峰最少,但大黃素的測量得率降低較多。為了減少損耗,降低實驗誤差,在該方法測定大黃素分離度好的情況下,筆者最終選擇乙酸乙酯萃取后蒸干加甲醇作為最終的供試品制作方法,以保留大黃素的最大含量。
根據《廣東省醫療機構制劑規范》中痔瘺寶洗劑鑒別法“乙醚萃取后蒸干”“殘渣加乙酸乙酯溶解”,筆者亦試用此方法作對比,結果采用乙酸乙酯溶解供試品的色譜峰出現嚴重的溶劑前沿,用甲醇溶解的液相色譜峰峰型對稱,幾乎沒有溶劑峰。由于直接使用甲醇較配制流動相方便,所以最終確定用甲醇溶解。
高效液相檢測流動相的選擇:筆者曾按2015 版《中華人民共和國藥典》中大黃含量測定方法(甲醇∶0.1%磷酸溶液=85∶15,C18色譜柱(安捷倫))試驗,結果大黃素在4.4 min 出峰,大黃蒽醌的最后一個色譜峰(大黃素甲醚)亦在7.5 min 出峰。由于中藥制劑成分復雜,出峰時間過早,容易與雜質峰重疊,影響分離度,故本流動相開始用降低甲醇的比例至50%來加快極性大的雜質出峰,推遲大黃中的蒽醌出峰,確保大黃素的分離度達到要求,3~5 min調整甲醇比例至70%亦是使雜峰與大黃蒽醌更好的分離;5~17 min 逐漸調整甲醇比例至86,使大黃蒽醌出峰分離,20~22 min 調整甲醇至50%使流動相比例恢復初始狀態,22~25 min 保持甲醇比例50%使基線達到平衡。從圖譜上看,大黃5 種蒽醌的出峰的初末時間差由原來“藥典方法”的5 min 變至10 min,除保留時間最短的蘆薈大黃素有一肩峰外,其余4 種蒽醌的分離度好,達到檢測要求。
本研究由肇慶高等醫學專科學校與肇慶市中醫院利用各自的優勢,聯合起來進行該課題的研究,根據廣東省食品藥品監督管理局《關于進一步加強醫療機構制劑配制監督管理工作的通知》要求,按《廣東省醫療機構制劑質量制訂的技術要求》,參照藥典和文獻,重新修訂其質量標準和工藝,使其達到《中國藥典》的先進水平,為保證該制劑的安全有效提供強而有力的技術支撐。
