氣相色譜法測定水果中多種有機磷農藥殘留分析
2019-06-11 05:31:53李恒毛林夏
安徽農業科學 2019年3期
李恒 毛林夏


摘要[目的]建立了水果樣品前處理,毛細管柱氣相色譜法(GC-FPD)測定水果中6種有機磷農藥含量的快速檢測方法。[方法]以6種有機磷農藥為檢測對象,通過超聲波系統輔助提取水果中農藥殘留,GC-FPD測定水果中6種有機磷農藥的含量。[結果]在0.025~0.500 μg/mL濃度范圍內,農藥殘留濃度和峰面積有很好的相關性,相關系數r>0.995,回收率為71.2%~104.5%,精密度(RSD)為1.66%~5.23%,方法檢出限低于國家標準,適合大批量樣品檢測,能夠大幅縮短樣品前處理時間,提高檢測效率。[結論]該方法樣品前處理過程簡單、處理速度快,對欲測定農殘組分的選擇性和回收率均能夠達到痕量分析要求,可以復制推廣至水果中其他有機磷農殘的檢測。
關鍵詞氣相色譜法;有機磷農藥;超聲波;殘留
中圖分類號S48 1+.8文獻標識碼A
文章編號0517-6611(2019)03-0193-03
doi:10.3969/j.issn.0517-6611.2019.03.060
有機磷農藥是指含磷元素的有機化合物農藥,是一類廣譜、高效的化學殺蟲劑[1],主要用于防治植物病、蟲、草害,所以其在農業生產中被廣泛使用,導致農作物產品中發生不同程度的殘留。有機磷農藥對人、畜的毒性較大,在農業生產中,如果不按規定的用藥量、次數、方法或安全間隔期施藥,就會引起農藥超標或農藥中毒,危害人類健康。世界各國都對食品中農藥殘留最大限量[2]進行了規定。現代農藥殘留分析方法通常包括樣品前處理和測定兩部分[3],樣品前處理是核心。農藥殘留的試樣前處理過程分為提取、濃縮、凈化等關鍵步驟[4]。高效快速的提取方法是農藥殘留分析的核心,它決定著分析的準確性和重現性,樣品前處理方法的選擇直接影響檢測的效率和最終分析結果,所以大力開展農藥殘留量檢測技術以及相關的前處理技術的研究,發展簡單、快速、有效的前處理方法是非常必要的,對于保護人民群眾舌尖上的安全具有重要意義。目前,有機磷的檢測主要有氣相色譜法(GC)[5-7]、氣相色譜質譜法(GC-MS)[8-9]和氣相色譜-質譜-質譜法(GC-MS/MS)[10]等。GC-MS和GC-MS/MS靈敏度較高,但是價格昂貴,運行成本高。氣相色譜法的主要特點是選擇性高、分離效率高、靈敏度高、分析速度快[11],被廣泛應用于食品安全中有機磷農藥殘留的檢測。筆者通過超聲波系統輔助提取水果中農藥殘留,毛細管柱氣相色譜法(GC-FPD)測定水果中6種有機磷農藥含量,建立水果樣品前處理檢測的快速方法。
1材料與方法
1.1主要儀器氣相色譜儀(Agilent 7890A,FPD檢測器,7683B自動進樣器,DB-17色譜柱);GM200盤式粉碎磨;電子天平;旋轉蒸發儀;渦旋混合器;超聲波清洗機(型號鼎泰恒盛DTC-27J);所有用到的計量器具均檢定合格。
1.2主要試劑敵敵畏、樂果、甲基對硫磷、毒死蜱、馬拉硫磷、殺撲磷標準儲備液(均為100 μg/mL,農業部環境保護科研監測所)。乙腈、丙酮、甲醇為色譜純;氯化鈉為分析純;試驗用水為GB/T 6682規定的 Ⅰ 級水。
1.3混合標準工作溶液的配制分別將裝有敵敵畏、樂果、甲基對硫磷、毒死蜱、馬拉硫磷、殺撲磷的標準品(100 μg/mL)的安瓿瓶從冰箱中(2 ℃)取出,在室溫下放置15 min后打開,分別將安瓿瓶中1 mL標準品全部轉入10 mL容量瓶中,用丙酮清洗安瓿瓶3次,定容至刻度,即得10 μg/mL的混合標準儲備溶液,采用逐級稀釋法將10 μg/mL混合標準儲備溶液配制成濃度分別為0.025、0.050、0.100、0.200、0.300、0.400、0.500 μg/mL的混合標準溶液。
1.4方法
1.4.1試驗原理。試樣中有機磷類農藥經乙腈提取,提取溶液經過濾、濃縮后,用丙酮定容,注入氣相色譜儀,農藥組分經色譜柱分離,用火焰光度檢測器(FPD)檢測。用色譜柱的保留時間定性,外標法定量[12]。
1.4.2試驗步驟。將水果樣品切碎,使用盤式粉碎磨制成勻漿。準確稱取10.0 g經過勻漿制備好的試樣于帶塞塑料瓶中,加入乙腈25 mL,加入2 g氯化鈉,置于超聲波恒溫清洗機中,將水溫調至20 ℃后超聲提取10 min后靜置,使乙腈和水相分層。吸取上層提取液于茄形瓶中,在45 ℃水浴減壓旋轉蒸發儀上蒸餾至近干,加入2.0 mL丙酮后在渦旋混勻器上充分清洗混勻后轉移至自動進樣器樣品瓶中,上機測試。
1.4.3提取條件的選擇。有機磷農藥在乙腈中有良好的溶解性,在超聲波加速反應系統輔助下提取殘留農藥,減小了樣品和提取液與容器接觸的面積,農藥殘留損失小。在超聲波輔助提取過程中,超聲波產生的沖擊波對樣品直接反復高頻沖擊,能破壞樣品和農藥的表面吸附,且超聲波產生的沖擊力能起到一定的攪拌作用,使樣品表面能不斷與新鮮的溶劑接觸,加速農藥在提取液中的溶解,同時使用一定的溫度,更是加快了樣品中殘留農藥的擴散速度。根據樣品的性質、提取效果、塑料瓶體積、提取時效和檢測成本等方面因素綜合考慮,最終確定用25 mL體積乙腈提取液,于20 ℃保持10 min進行農藥殘留提取。該提取方法實際使用效果良好,大大縮短了水果中農殘提取的時間,加快了分析的速度。
1.4.4樣品濃縮過程條件的控制。在做回收率試驗時發現,通過旋轉蒸發法對農殘提取液進行濃縮過程中,如果水浴溫度過高和濃縮至干后定容都會使大多數農藥的回收率明顯偏低,這可能是溫度過高引起有機磷農藥的分解,溶劑揮干后使有機磷分解或蒸干的緣故,所以在試驗過程中,水浴溫度不宜過高,控制在45 ℃,提取液濃縮至近干即可。
1.4.5色譜分析條件。改變色譜柱、程序升溫條件及載氣流速、氫氣與空氣比例,考察待測農藥色譜分離情況及信號強度。色譜柱、程序升溫、載氣流速和氫氣與空氣比例不同,待測農藥的分離情況及信號強度也不同。根據在最短時間獲得最佳分離效果的原則,得到如下色譜分析條件:氣相色譜柱DB-17(30 m×0.25 mm×0.25 μm),載氣為氮氣
(99.999%),燃燒氣體為高純氫氣和空氣(99.999%,氫氣與
空氣比例為75∶100);流速為4 mL/min,進樣方式為不分流進樣;進樣口溫度230 ℃,FPD檢測器溫度250 ℃,柱溫由150 ℃
(保持2 min)升至250 ℃(8 ℃/min),保持6 min,進樣量1 μL。
2結果與分析
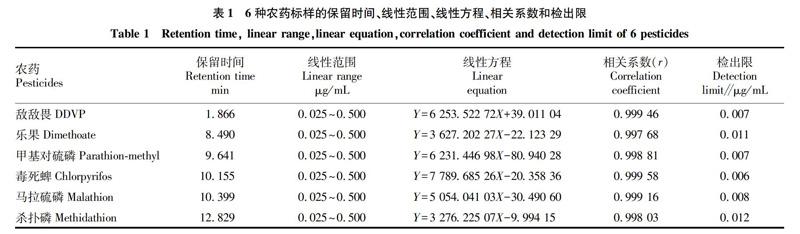
2.1系統適應性試驗按照“1.4.5”色譜分析條件,對敵敵畏、樂果、甲基對硫磷、毒死蜱、馬拉硫磷、殺撲磷6種有機磷農藥殘留混合標準溶液進行上機測試,待測農藥分離情況見圖1。從圖1可以看出,在“1.4.5”條件下,混合標準品色譜圖中敵敵畏、樂果、甲基對硫磷、毒死蜱、馬拉硫磷、殺撲磷6種有機磷農藥殘留混合標準溶液的保留時間分別為1.866、8.490、9.641、10.155、10.399、12.829 min,其色譜峰與相鄰色譜峰分離度良好(與相鄰峰的分離度均大于1.5),符合氣相色譜法定量分析的要求。
2.2線性范圍、相關系數和檢出限準確配制0.025~0.500 μg/mL不同濃度的各種有機磷標準品工作溶液系列,按照“1.4”中方法和條件進行測定,以峰面積對濃度做線性回歸分析,繪制標準曲線,得到待測農藥的線性方程及相關系數,檢出限按信噪比3∶1 計算,結果見表1。從表1可以看出,敵敵畏、樂果、甲基對硫磷、毒死蜱、馬拉硫磷、殺撲磷6種有機磷農藥在0.025~0.500 μg/mL線性關系良好,說明峰面積和濃度之間有很好的相關性,能夠達到實驗室快速檢測的要求,滿足進出口水果農藥殘留檢測分析的要求。
2.3方法的加標回收率和精密度試驗為了考察該方法的回收率和精密度,以桔子樣品為背景分別添加3個水平的混合標準品溶液,按照“1.4.5”方法進行提取濃縮測定,每個添加水平做7次平行試驗,計算各待測組分的回收率和精密度(RSD)。結果表明(表2),6種有機磷類農藥的加標回收率為71.2%~104.5%,精密度(RSD)為1.66%~5.23%,回收率和精密度均符合痕量分析的要求,能夠滿足實驗室對進出口水果有機磷農殘的檢測需求。
2.4水果樣品檢測應用該試驗所建立的處理方法和條件,對實驗室常檢樣品葡萄、石榴、蘋果、梨和桔子中6種有機磷進行檢測,結果顯示,葡萄、蘋果和梨中毒死蜱有檢出,毒死蜱檢測結果均在食品中農藥最大殘留限量標準允許范圍,其他5種農藥殘留無檢出,石榴和桔子中均無這6種有機磷農藥殘留檢出。
3結論
該研究通過超聲波系統輔助提取水果中農藥殘留,毛細管柱氣相色譜法(GC-FPD)測定水果中6種有機磷農藥含量。此方法在水果樣品浸漬提取的同時,輔以超聲波提取,在合適的溫度下完成樣品的前處理工序,操作簡單,提取效果好,快速節能、節省樣品前處理的溶劑,污染小,可同時處理多組樣品,省時高效,顯著提高了樣品前處理效率,進一步提高了實驗室檢測工作效率,適合進出口水果中有機磷農殘的快速檢測工作,能夠更好地為檢驗檢疫執法提供技術支持。
參考文獻
[1] 吳海燕,陳建軍.蔬菜中多種有機磷農藥殘留氣相色譜分析法[J].食品與機械,2010,26(4):76-77,83.
[2] 中華人民共和國國家衛生和計劃生育委員會,中華人民共和國農業部,國家食品藥品監督管理總局.食品中農藥最大殘留限量:GB 2763—2016[S].北京:中國標準出版社,2017.
[3] 唐波,張金娥,齊力匯,等.氣相色譜法測定農藥殘留量新進展[J].山東師范大學學報(自然科學版),2002,17(4):35-39.
[4] 張福金,劉建平,馮小慧,等.氣相色譜法檢測蔬菜和水果多組分農藥殘留中若千問題的研究[J].內蒙古農業科技,2006(3):31-32.
[5] 陸小磊,吳慧明,金紹強,等.毒死蜱在4種作物中殘留量的氣相色譜分析方法[J].浙江農業科學,2009(1):168-172.
[6] 陸勛元,蔣永祥.氣相色譜法同時檢測和分析蔬菜中毒死蜱、氯氰菊酯和氰戊菊酯的殘留量[J].理化檢驗(化學分冊),2006,42(9):737-739.
[7] 喻龍,李光義,鄧曉,等.蔬菜中殘留毒死蜱的檢測方法[J].中國農學通報,2007,23(12):338-340.
[8] 汪軍.GC-MS定性定量分析出口西蘭花樣品中毒死蜱的農藥殘留量[J].現代農藥,2002(1):23-24.
[9] 徐穎潔.氣相色譜-質譜法測定農藥中14種有機氯·有機磷成分[J].安徽農業科學,2014,42(17):5470-5471,5646.
[10] 凌云,王菡,雍煒,等.氣相色譜-質譜/質譜法檢測蔬菜中的毒死蜱及其代謝物[J].色譜,2009,27(1):78-81.
[11] 于世林,杜振霞.化驗員讀本:下冊(儀器分析)[M].北京:化學工業出版社,2004.
[12] 中華人民共和國農業部.蔬菜和水果中有機磷、有機氯、擬除蟲菊酯和氨基甲酸酯類農藥多殘留的測定:NYT 761—2008[S].北京:中國農業出版社,2008.
