茶葉中蒽醌的內標-氣相色譜-串聯質譜法測定
2019-06-17 07:16:38高慧汪洋邢燕王勤尚萌琳
食品研究與開發 2019年12期
高慧,汪洋,邢燕,*,王勤,尚萌琳
(1.淄博市疾病預防控制中心,山東 淄博 255026;2.山東大學公共衛生學院,山東 濟南 250000)
中國是全球較早的茶葉出產和銷售的國家,也是全球最大的茶葉消費國,隨著經濟全球化發展,茶葉的出口量也持續增加。蒽醌在自然界中廣泛存在,茶葉中蒽醌的殘留污染可能來源于農藥殘留或產品包裝。因歐盟認為蒽醌具有致癌性,2014年10月,歐盟新發布的(EU)No1146/2014 中規定了茶葉中蒽醌的最高限量為0.02 mg/kg[1]。我國尚無最高限量要求。2014年我國茶葉出口同比下降。中國輸歐茶葉因不符合限值要求被通報27 次,其中蒽醌8 次,居首位[2]。研究茶葉中蒽醌的污染來源和控制技術,降低茶葉中蒽醌污染物含量,破除歐盟技術壁壘是一個急需解決的問題,因此,建立茶葉中蒽醌的定量檢測方法至關重要。
9,10-蒽醌,又名蒽醌(anthraquinone,CAS No.84-65-1),是一種黃色針狀結晶,易溶于熱苯、熱甲苯、乙醇、乙醚和氯仿,不溶于水,蒽醌常存在于高等植物和低等植物地衣類和真菌類的代謝產物中,主要用于染料生產的中間體,是媒染染料、酸性染料和還原染料的主要原料。還用做紙制品印花的導氧劑。GB/T 23204-2008《茶葉中519 種農藥及相關化學殘留量的測定氣相色譜-質譜法》[3]涉及蒽醌殘留量的測定,但為定性檢測方法,不適合于茶葉中微量蒽醌的測定。目前報道的蒽醌類定量測定的方法主要有分光光度法[4-7]、高效液相色譜法[8-10]、氣相色譜-質譜聯用法[11]、氣相色譜-串聯質譜法[12-16]、高效毛細管電泳法[17]等。其中分光光度法和高效液相色譜法多為測定總蒽醌含量,不適合于茶葉中蒽醌的測定;氣相色譜-質譜法和氣相色譜-串聯質譜法多為凝膠聯合凈化或SPE 小柱凈化,能夠較好的純化分離出目標物,但凈化過程比較繁瑣,耗時較長;高效毛細管電泳法普及率比較低。本研究通過對樣品前處理條件、色譜條件等的優化,建立了一種準確、快速測定茶葉中蒽醌的氣相色譜-串聯質譜法(gas chromatography-tandem mass spectrometry,GC-MS/MS)內標分析方法。
1 材料與方法
1.1 儀器與試劑
Agilent 7010 三重四極桿氣質譜聯用儀[配電子轟擊離子源(EI)]:美國 Agilent 公司;N-EVAP 112 氮吹濃縮儀:美國 OA-SYS 公司;CP-225D 電子天平:德國Sartorius 公司;ST-16R 高速冷凍離心機:美國Thermo公司;KS-500D 型超聲波清洗器:寧波科生儀器廠;Milli-Q 超純水系統:美國Millipore 公司。N-丙基乙二胺(Bondesil-PSA,40 μm,100 gm)、Bondesil-C18(40 μm,100 mg):美國 Agilent 公司。無水硫酸鈉(AR 級):國藥集團化學試劑有限公司;乙腈(HPLC 級)、丙酮(HPLC級)、環己烷(HPLC 級)、乙酸乙酯(HPLC 級):德國Merck 公司;蒽醌(純度≥98.0%):上海安譜公司;蒽醌-D8 標準物質(純度>99.0%):美國TRC 公司。
1.2 標準工作液的配制
稱取0.010 0 g 蒽醌,加丙酮超聲、溶解、稀釋定容至10 mL,濃度為1 000 μg/mL,即為蒽醌標準儲備液。取蒽醌標準儲備液10 μL,加環己烷-乙酸乙酯(1+1)至 10 mL,即為蒽醌應用液,濃度為 1 μg/mL,于 0 ℃~4 ℃冰箱內避光保存。稱取0.010 0 g 蒽醌-D8 標準物質,加丙酮超聲、溶解、稀釋定容至10 mL;再取100 μL,加環己烷-乙酸乙酯(1+1)至 1 mL,即為蒽醌-D8 內標應用液,濃度為 100 μg/mL,于 0 ℃~4 ℃冰箱內避光保存。
1.3 試驗方法
1.3.1 色譜條件
色譜柱:HP-5MS(30 m×250 μm,0.25 μm);載氣:高純氦氣,純度>99.999%,恒流模式,進樣口溫度:280 ℃;流速1.0 mL/min;程序升溫:初溫100 ℃,保持1 min,以 10 ℃/min 升到 200 ℃,保持 2 min,繼續以 25 ℃/min升到 280 ℃,保持 10 min;進樣量:1 μL;進樣方式:不分流進樣。
1.3.2 質譜條件
電離方式:電子轟擊離子源(EI);離子源溫度:230 ℃;接口溫度:300 ℃;電子能量 70 eV;碰撞氣:高純氬氣,純度>99.999%,;溶劑延遲:3.75 min;掃描方式:多反應監測模式(multiple reaction monitoring,MRM)掃描模式;駐留時間:0.10 s。標準的保留時間、定性離子、定量離子對等具體質譜參數見下表1。

表1 標準的質譜參數表Table 1 The mass spectrometry parameters of standards
1.3.3 樣品前處理
將100 g 茶葉用粉碎機粉碎,混勻,分裝于兩個50 mL 潔凈棕色玻璃瓶中,一份用于遮光密閉保存留樣,另外一份用于檢測。
1.3.3.1 樣品提取
精確稱取2 g(精確至0.001 g)粉碎、混勻茶葉樣品于50 mL 離心管中,加水3 mL,加蒽醌-D8 內標應用液 10 μL,渦旋 30 s 潤濕樣品,加 10 mL 乙腈,超聲提取30 min,加無水硫酸鈉2 g,渦旋混勻1 min,以8 000 r/min 離心10 min。上清液待凈化。
1.3.3.2 樣品QuEChERS 凈化
取乙腈提取液約8 mL,加入0.8 g 無水硫酸鈉,0.8 g PSA 粉末和0.4 g C18 固相吸附劑,渦旋1 min,8 000 r/min 離心 5 min。取上清液 5.0 mL,40 ℃氮吹濃縮至近干,加1.0 mL 乙腈溶解殘渣,渦旋1 min,以8 000 r/min 離心 5 min,上清液過 0.22 μm 聚四氟乙烯過濾膜,待測定。
1.3.4 標準曲線繪制
分別準確吸取1.2 中蒽醌標準應用液(1 μg/mL)10、20、50、100、150、200 μL 于 1 mL 樣品瓶中,分別各加入蒽醌-D8 內標應用液(100 μg/mL)5 μL,用乙腈定容至 1 mL,蒽醌-D8 的濃度為 0.50 μg/mL,蒽醌標準系列溶液濃度為 10、20、50、100、150、200 ng/mL。以蒽醌的濃度為橫坐標,定量離子對(208>180)的峰面積與內標峰面積的比值為縱坐標繪制標準曲線。標準總離子流圖見圖1。
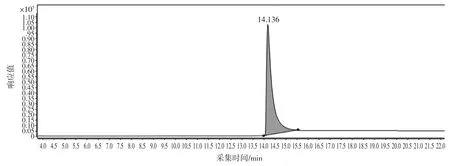
圖1 標準總離子流圖Fig.1 The total ion chromatogram of standard
2 結果與討論
2.1 色譜條件的選擇與優化
由于茶葉中基質復雜,測定茶葉中蒽醌必須選擇能夠很好的把目標物與干擾物分離的色譜柱,試驗比較了 HP-5MS、DB-5MS、DB-1701P 3 種不同的色譜柱,DB-5MS、DB-1701P 和 HP-5MS 3 種色譜柱的樣品加標色譜圖見圖2、圖3、圖4。
改變色譜分離條件,觀察分離情況,不同色譜柱對目標物的分離情況見下表2。
表2 結果表明,HP-5MS 較其他兩種色譜柱分離效果更好。
進樣口溫度過高,會致使蒽醌分解,溫度過低,蒽醌汽化不完全,靈敏度均會下降。考查了240 ℃到290 ℃之間的進樣口溫度,不同進樣口溫度對蒽醌峰面積的影響見圖5。
由圖5 發現溫度在280 ℃時,蒽醌氣化完全,未見分解。
2.2 質譜條件的選擇與優化
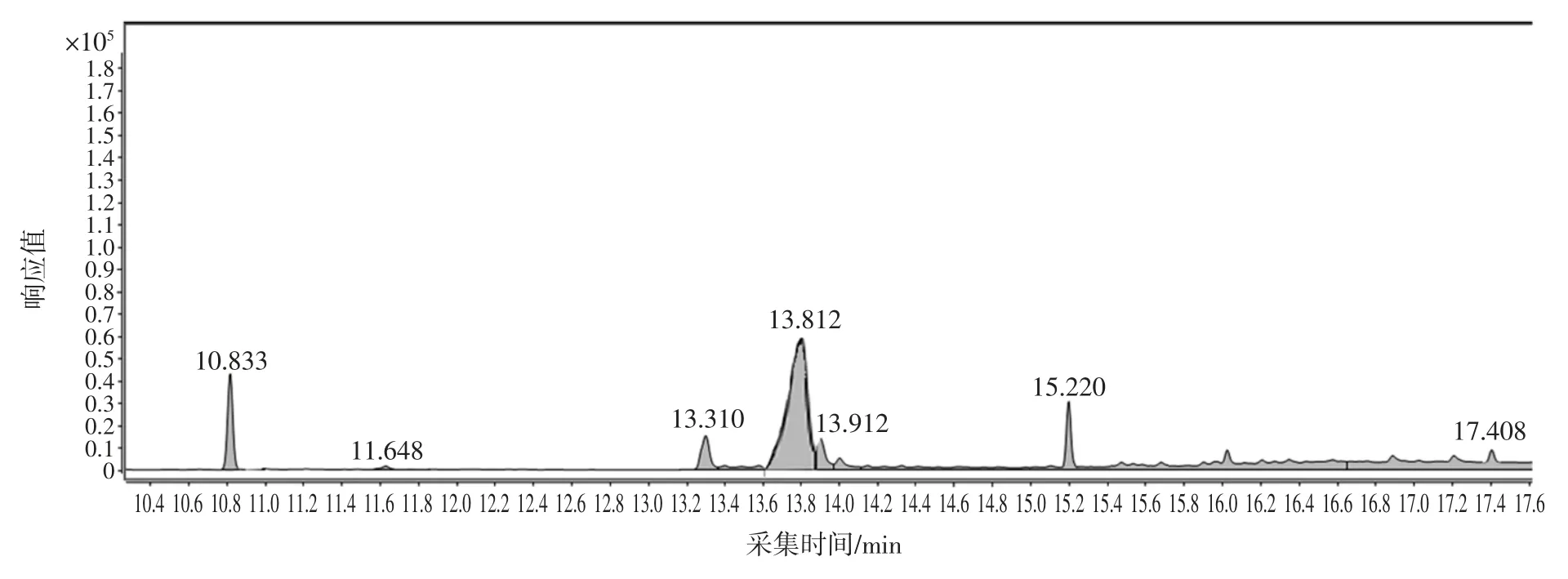
圖2 DB-5MS 樣品加標色譜圖Fig.2 The total ion chromatogram of adding sample separating by DB-5MS chromatographic column
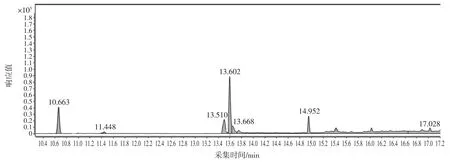
圖3 DB-1701 樣品加標色譜圖Fig.3 The total ion chromatogram of adding sample separating by DB-1701 chromatographic column
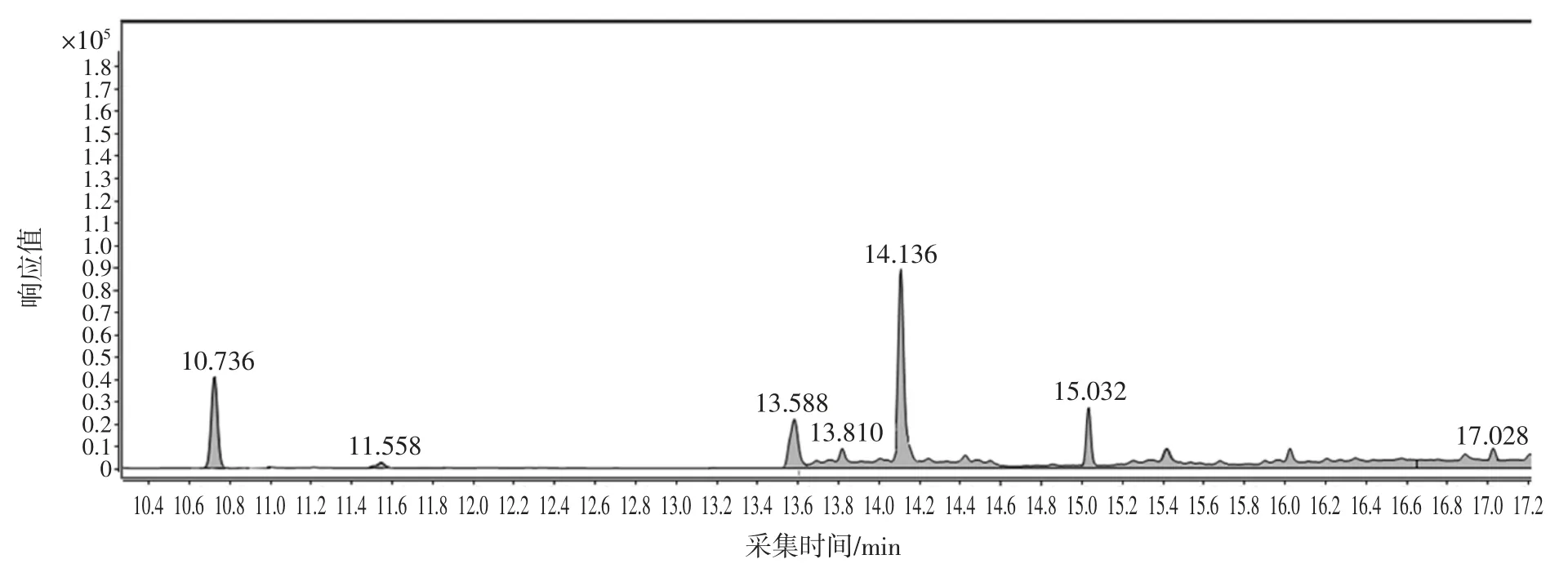
圖4 HP-5MS 樣品加標色譜圖Fig.4 The total ion chromatogram of adding sample separating by HP-5MS chromatographic column
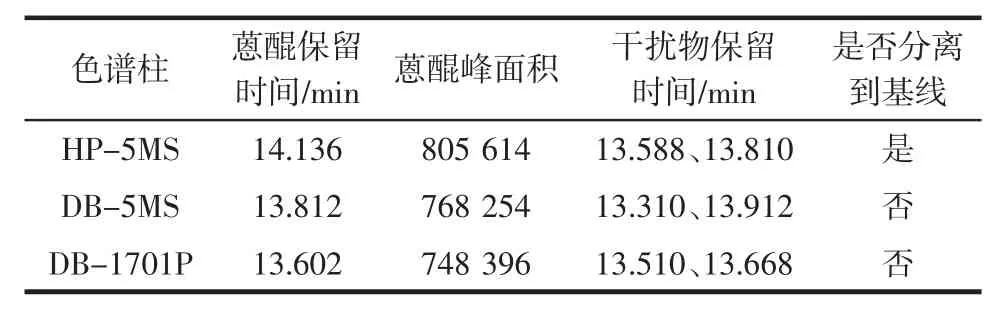
表2 不同色譜柱的分離情況Table 2 The separation results of adding sample by different chromatographic columns
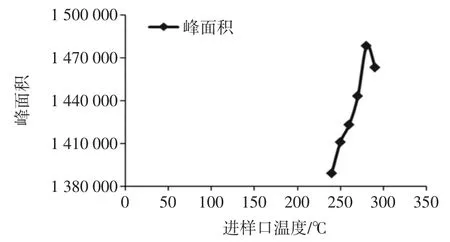
圖5 不同進樣口溫度對蒽醌峰面積的影響Fig.5 The effect of different inlet temperatures on anthraquinone peak area
在優化的色譜條件下進行全掃描(Scan),得到蒽醌的質譜圖,選擇質核比較大,相對豐度高的離子作為產物離子。再用MRM 方式掃描,為保證掃描結果的準確度,按待測組分保留時間分組,使待測物有充足的時間掃描。分別選擇了豐度較高的兩組離子m/z 208,180,m/z 208,152 為定量和定性離子對,對產物離子進行掃描。為提高檢測的靈敏度,根據離子信息,選取不同的碰撞能量觀察離子對的響應情況,最終選取了10 eV 做為定量和定性離子對的碰撞能量。
2.3 樣品的基質效應
氣相色譜串聯質譜法測定樣品時,有時基質對待測物的離子具有抑制或增強效應,因此選用內標法定量以提高靈敏度和準確性。分別制備用空白基質樣品溶液和乙腈稀釋的蒽醌和蒽醌-D8 標準工作溶液,對空白茶葉樣品進行低、中、高3 個濃度的添加水平試驗,用內標法進行定量,乙腈配制與空白基質樣品溶液配制的工作曲線回收率沒有明顯差異,比較接近,故選用了乙腈配制標準工作液。不同溶劑蒽醌的加標回收率和相對標準偏差見表3。
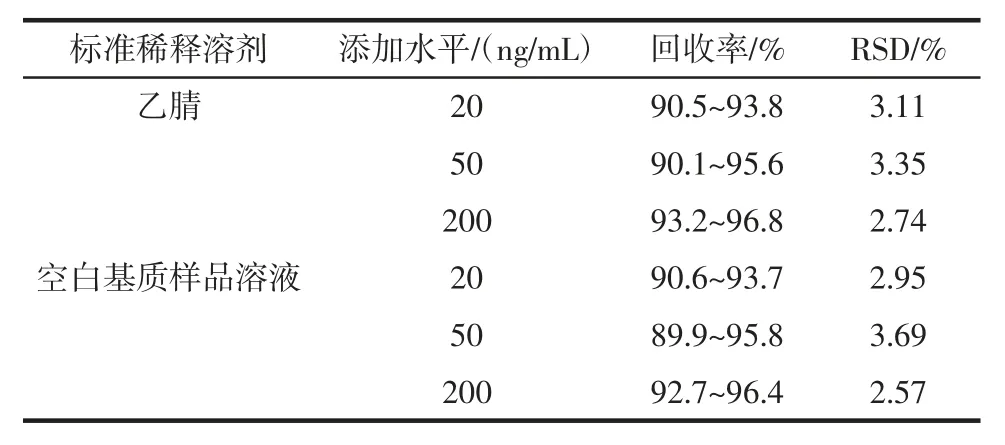
表3 不同溶劑蒽醌的加標回收率和相對標準偏差Table 3 The recovery rate and relative standard deviation(RSD)of anthraquinone in different solvents
2.4 樣品前處理條件的優化
2.4.1 提取效應
為了測試茶葉樣品加水后檢測,對檢測結果有無影響。試驗稱取了8 份蒽醌陽性茶葉樣品,其中4 份加水3 mL,渦旋30 s 潤濕樣品,另4 份不加水,分別加入10 mL 乙腈,后續步驟按照1.3.3.1 和1.3.3.2 提取、凈化。試驗數據顯示,茶葉樣品加水潤濕后再加溶劑提取、凈化,回收率更高。結果見表4。
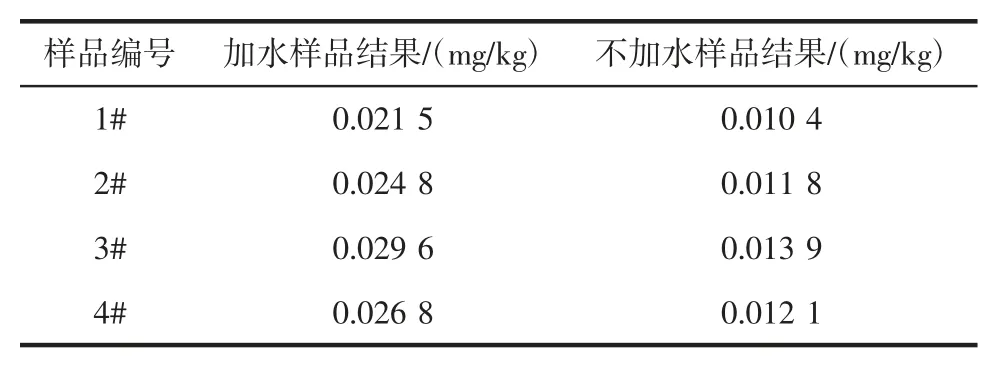
表4 樣品測試結果比較Table 4 The comparison of sample results
2.4.2 樣品凈化的選擇
分別選用了SPE 小柱和QuEChERS 兩種凈化方式進行比較。首先選用了TPC(ProElut TPC-2 固相萃取柱,6 mL,迪馬公司)小柱填充物上加入1 g 無水硫酸鈉,用 10 mL 乙腈-甲苯(3+1,體積比)活化;乙腈提取液 5.0 mL 上樣,棄去流出液;用 8 mL 甲苯-乙腈(1+3,體積比)淋洗,棄去流出液;再用12 mL 甲苯-乙腈(1+3,體積比)洗脫,抽干,收集洗脫液。40 ℃氮吹濃縮至近干,加 1.0 mL 乙腈溶解殘渣,渦旋 1 min,8 000 r/min 離心5 min,上清液過0.22 μm 聚四氟乙烯過濾膜,待測定。QuEChERS 凈化方式一:取乙腈提取液約8 mL,加入0.8 g 無水硫酸鈉,0.8 g 石墨化炭黑粉末和0.4 g C18 固相吸附劑,渦旋 1 min,8 000 r/min 離心 5 min。取上清液5.0 mL,40 ℃氮吹濃縮至近干,加1.0 mL 乙腈溶解殘渣,渦旋1 min,以8 000 r/min 離心5 min,上清液過0.22 μm 聚四氟乙烯過濾膜,待測定。QuECh-ERS 凈化方式二同1.3.3.2。不同凈化方式對蒽醌回收率的影響見圖6。
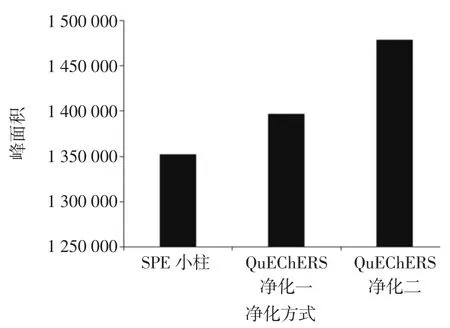
圖6 不同凈化方式對蒽醌回收率的影響Fig.6 The effect of different purification ways on the recovery rate of Anthraquinone
圖6 檢測結果表明,選用QuEChERS 凈化方式二凈化效果較好和回收率最高,因此選用了1.3.3.2 QuEChERS 凈化方式。
2.5 線性范圍及檢出限
在本試驗條件下,按照1.3 的試驗條件進行測定,以蒽醌的濃度為橫坐標,定量離子對(208>180)的峰面積與內標峰面積的比值為縱坐標繪制標準曲線。結果顯示:蒽醌在10 ng/mL~200 ng/mL 范圍內線性關系良好。同時做空白樣品加標試驗,以3 倍信噪比計算檢出限(limit of detection,LOD),以信噪比 10 倍計算定量限(limit of quantitative determination,LQD),結果見表5。

表5 蒽醌的線性方程、線性范圍、相關系數、LOD、LQDTable 5 The linear equation,linear range,correlation coefficient,LOD,LQD of anthraquinone
2.6 加標回收率和精密度
選取了未檢出蒽醌的綠茶、紅茶、烏龍茶、白茶4種樣品,分別稱取2 g(精確至0.001 g)樣品于50 mL離心管中,分別添加質量濃度為20、50、200 ng/mL 的標準溶液,每個添加水平取6 份平行樣,充分混勻后,按照最佳前處理和色譜質譜分析條件進行測定,回收率在 87.1%~98.4%之間,RSD 為 1.23%~4.12%,結果見表6。茶葉樣品總離子流圖見圖7,茶葉樣品加標總離子流圖見圖8。
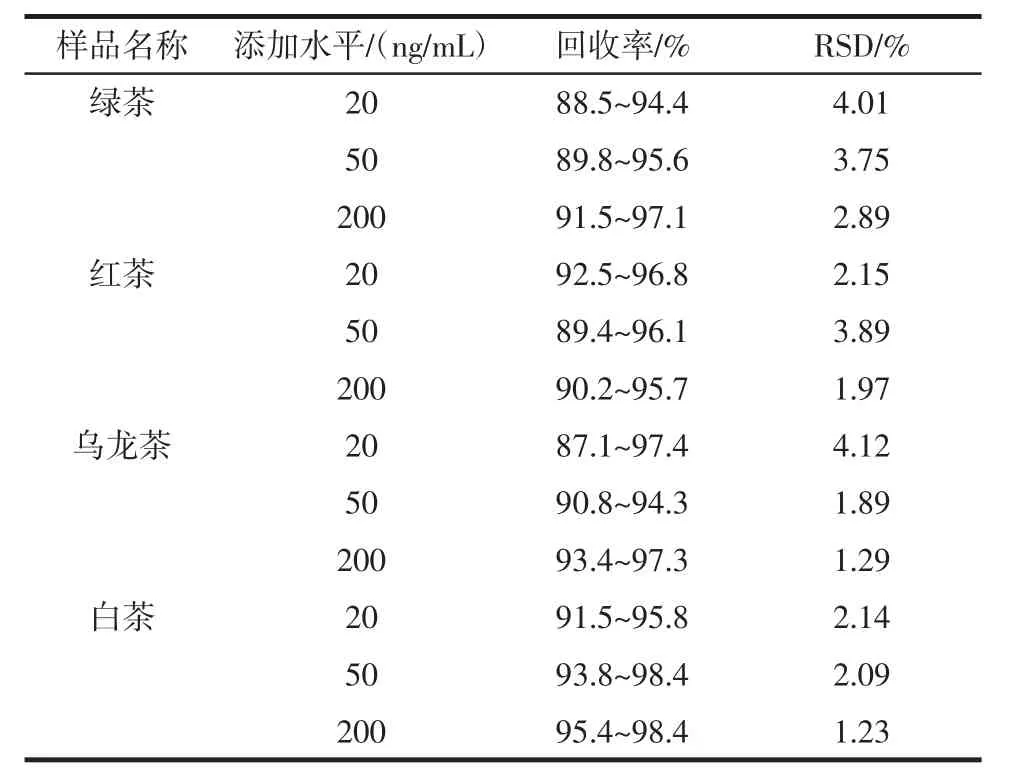
表6 蒽醌的加標回收率和相對標準偏差Table 6 The recovery and relative standard deviation(RSD)of anthraquinone
2.7 樣品測定
隨機采集市售茶葉50 份,包括綠茶15 份、紅茶15 份、烏龍茶10 份、白茶10 份。其中蒽醌檢出12 份,檢出率24.0%,其中紅茶檢出率最高為33.3%,白茶檢出率最低為13.3%;超標8 份,超標率為16.0%,其中、綠茶 2 份、紅茶 3 份、烏龍茶 2 份、白茶 1 份。通過測定結果可以看出,茶葉中的蒽醌確有檢出。該方法適用于茶葉中蒽醌的測定。
3 結論
本研究通過對茶葉樣品的前處理條件和色譜質譜條件的優化,選擇了最佳檢測條件,實現了對茶葉中蒽醌的測定,達到了很好的分離效果。該方法具有操作簡便、線性范圍寬、檢出限低、靈敏度高、雜質干擾少及結果準確可靠等優點。因茶葉中干擾物復雜,本文選用了QuEChERS 凈化方式,選用了無水硫酸鈉、
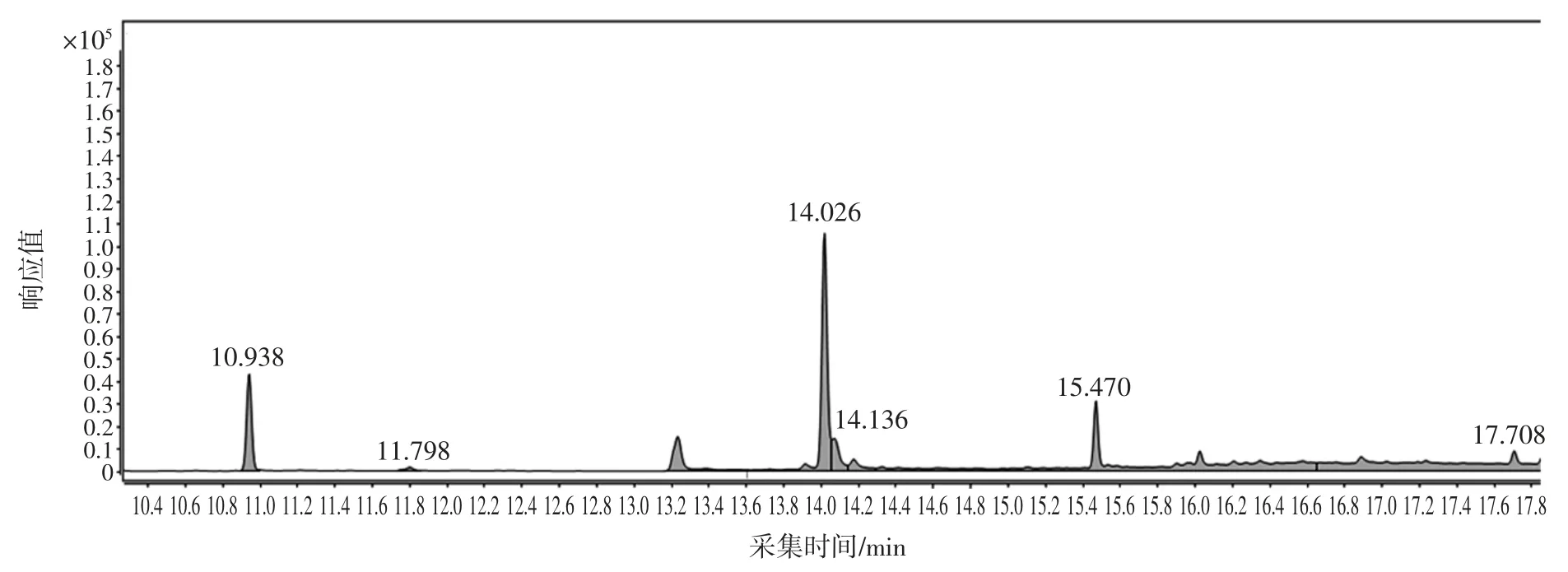
圖7 茶葉樣品的總離子流圖Fig.7 The total ion chromatogram of tea sample
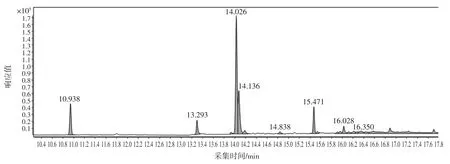
圖8 茶葉樣品加標的總離子流圖Fig.8 The total ion chromatogram of adding tea sample
PSA 粉末和C18 固相吸附劑,既能實現茶葉中復雜干 擾物的去除又能實現待測物的完全保留和洗脫,實現了氣相色譜串聯質譜法對茶葉中蒽醌的定量檢測,滿足了茶葉中蒽醌的分析測試要求。
