中老年獼猴單個核細胞miRNA表達差異及其靶基因預測
2019-06-27 02:53:34朱向情蔡學敏李自安龐榮清潘興華
西南國防醫藥 2019年6期
朱向情,王 強,蔡學敏,李自安,龐榮清,潘興華
衰老是機體細胞內基因結構、表達譜及表達水平、基因表達調控機制發生改變,導致生物活性分子、組織細胞及其組成成分、組織結構以及器官功能逐漸退化的生理過程。關于衰老發生學說,有體細胞突變、自由基、差錯災難、脂褐素累積、內分泌功能減退等[1],這些學說從不同角度解釋了衰老發生的原因,但均存在片面性。最近研究發現,機體免疫功能降低或紊亂、慢性炎癥是機體衰老的重要原因[2],但調控機制尚不清楚。外周血單個核細胞是機體免疫應答和防御的主要細胞組分,miRNA是一種含有約22個核苷酸的非編碼小分子RNA,不具編碼功能,但能夠與靶mRNA結合并調控基因表達和細胞生物進程[3]。miRNA對衰老相關基因表達也具有調控作用,已發現了mir-499、mir-34c等衰老調節miRNA,但數據十分有限[4]。本研究采用二代測序生物信息分析技術,研究了中年與老年獼猴外周血中單個核細胞的miRNA的表達差異及其靶基因,為揭示衰老發生機制提供新的科學數據。
1 材料與方法
1.1 實驗動物 選取8周歲和20~21周歲雌性健康獼猴各3只,分別設為中年組、老年組。每只采集外周血3.0 ml,用于單個核細胞分離和miRNA提取。所有獼猴均由中國科學院昆明動物學研究所動物中心提供,均飼養于醫院實驗動物中心,動物使用經過醫院實驗動物倫理委員會的批準,所有動物在普通級環境下、按照常規條件飼養。
1.2 miRNA測序及數據預處理 miRNA測序委托上海歐易生物醫學科技有限公司完成,使用Illumina公司Hiseq 4000測序儀測序。對測序產生的原始Fastq格式文件數據進行引物與載體序列去除、測序片段堿基的質量檢驗和長度篩選等,篩選高質量的測序片段。數據預處理通過perl腳本完成。
1.3 miRNA的識別 經過數據預處理后,根據Read長度統計片段長度分布情況,長度在21~25 nt的Read視為預測的miRNA。采用Bowtie軟件[5]將miRNA序列與miRBase數據庫中的miRNA進行比對,比對上的序列為已知miRNA。將未比對上的序列與Rfam數據庫中的序列進行比對,去除可能存在的其他類型的小RNA,將過濾后的序列與猴轉錄組序列進行比對,去除由降解mRNA測序產生的序列。將未同轉錄組序比對上的序列與RepBase數據庫進行比對,去除重復序列。剩余序列采用miRDeep2軟件[6]結合猴同源miRNA序列以及RNAfold軟件預測的二級結構,識別新的miRNA。
1.4 miRNA差異分析 比對已知的miRNA序列,進行表達量統計以及差異分析,miRNA表達量計算采用TPM計算度量指標,TPM是指以每百萬比配成對序列做miRNA表達量指標,利用DESeq2軟件包[7]中的負二項分布檢驗計算基因差異表達量。采用NB(負二項分布檢驗的方式)對reads數進行差異顯著性檢驗,根據P<0.05篩選出差異表達miRNA。
1.5 差異miRNA靶基因預測 對差異表達分析得到的miRNA,利用miranda算法預測miRNA靶基因。miranda算法是基于miRNA-3'UTR序列匹配與能量穩定性評估計算步驟綜合預測miRNA靶基因,該算法采用動態規劃算法搜索miRNA與3'UTR互補同時穩定形成雙鏈的區域,預測miRNA靶基因過程中所使用到的閾值參數為:S>=150,ΔG<=-30 kcal/mol和 Demand strict 5'seed pairing。其中S指匹配區域內single-residue-pair match scores;ΔG是雙鏈形成時的自由能。
1.6 預測靶基因GO和KEGG富集分析 利用Bioconductor(3.4 版本)(http://www.bioconductor.org/)項目中的GO和KEGG富集分析軟件包clusterProfiler(3.2.10版本),對預測到的差異miRNA靶基因進行GO和KEGG富集分析。
2 結果
2.1 差異表達miRNA 共發現差異表達miRNA 55個,其中21為已知miRNA(表1),在這些已知差異表達miRNA中,表達上調的有4個,分別為miR-449a-5p、miR-135a-1-3p、miR-9-5p 和 miR-615-3p;表達下調的有17個。發現新的差異表達的miRNA 34個,其中上調和下調miRNA分別為11個和23個,這些新miRNA的作用靶基因上不清楚。根據P值大小,前10個差異表達miRNA的信息見表2。
2.2 差異miRNA的靶基因預測 對21個已知差異miRNA進行靶基因預測,共找到1634個靶基因,其中值最小的miR-1262-3p預測到3個靶基因,分別為:VPS18 (基因登錄號:NM_001257653.1)、FGF14(基因登錄號:XM_015121402.1)和 FAM167A(基因登錄號:XM_015144755.1)。
2.3 靶基因GO分析 對P≤0.05的已知miRNA進行靶基因預測,發現其作用靶基因有1492個。進一步對靶基因進行GO聚類分析,證明這些基因主要設計生物進程、細胞組份和分子功能方面。與生物進程相關的靶基因主要涉及轉錄及調控、細胞凋亡、信號轉導、細胞分化等(圖1A),與細胞組份相關的靶基因主要涉及細胞膜、核、質膜、細胞液等(圖1B),與分子功能相關的靶基因主要涉及與ATP結合、DNA結合、RNA結合分子等(圖1C)。
2.4 miRNA靶基因KEGG通路富集分析 對靶基因進行KEGG通路富集分析,發現這些靶基因主要富集在35條信號通路上,其中前20個顯著富集的KEGG通路見圖2,所涉及的KEGG通路主要包括胰島素分泌、MAPK、PI3K-Akt、wnt、mTOR 信號通路及與細胞凋亡信號通路和免疫相關的toll樣受體信號通路等。
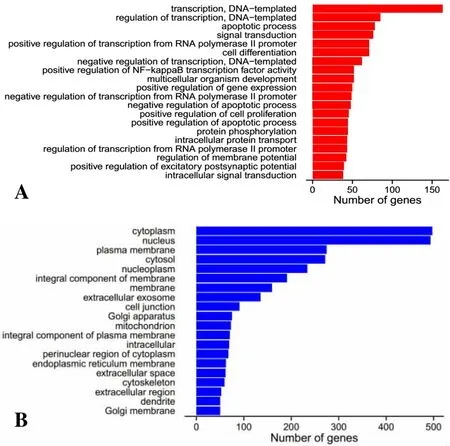
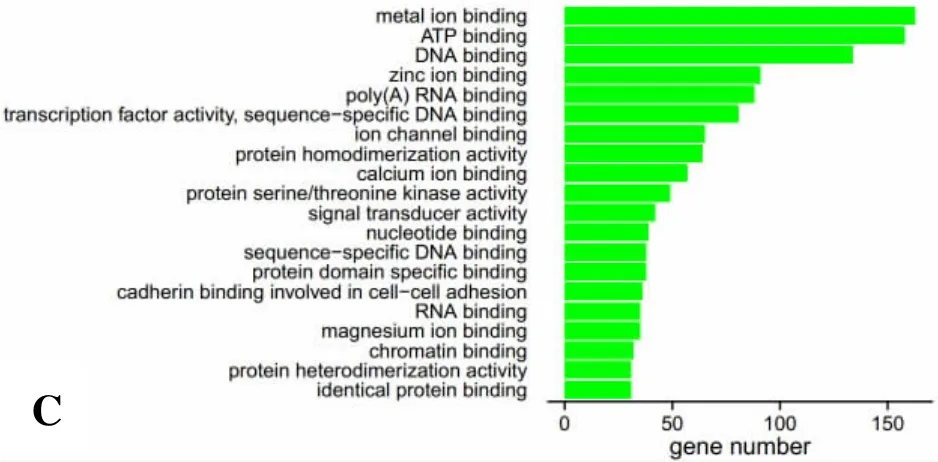
圖1GO聚類分析顯示顯著富集的前20個GO term柱狀圖
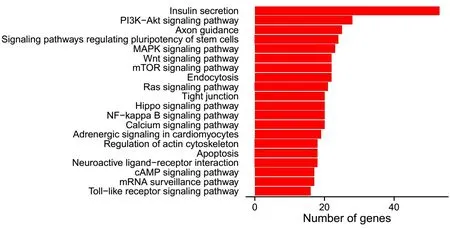
圖2 富集的前20個KEGG通路柱狀圖
3 討論
miRNA是機體中參與衰老調控的一類重要分子,其調控方式主要是通過堿基配對結合到衰老相關基因的mRNA的3'末端非編碼區,影響mRNA的翻譯,進而影響衰老進程[3,8]。有研究報道,mir-499、mir-34c、mir-1285、mir-34a 等 miRNA 能夠通過與靶基因結合影響細胞衰老進程[8]。本研究共發現55個在中年與老年獼猴單個核細胞中有顯著差異表達的miRNA,其中21個miRNA已被注釋,上調的 4個 miRNA分別為 miR-135a-1-3p、miR-449a-5p、miR-615-3p和miR-9-5p,提示這些下調或上調miRNA的差異表達參與了機體衰老進程。進一步55個差異表達的miRNA共調控1634個靶基因。對這些靶基因進行GO分析結果顯示,這些靶基因的作用主要涉及生物進程、細胞組份及ATP、DNA、RNA結合功能等;KEGG富集結果顯示,這些靶基因主要被顯著富集在胰島素分泌、wnt以及mTOR等與衰老相關的通路上,主要調節細胞增殖、分化和衰老等[9]。本研究發現的miRNA可以通過調控這些通路中的關鍵基因表達來促進或延緩細胞衰老,可為臨床上衰老相關疾病的診斷提供新的候選因子,也為衰老相關疾病的治療提供新的靶點。
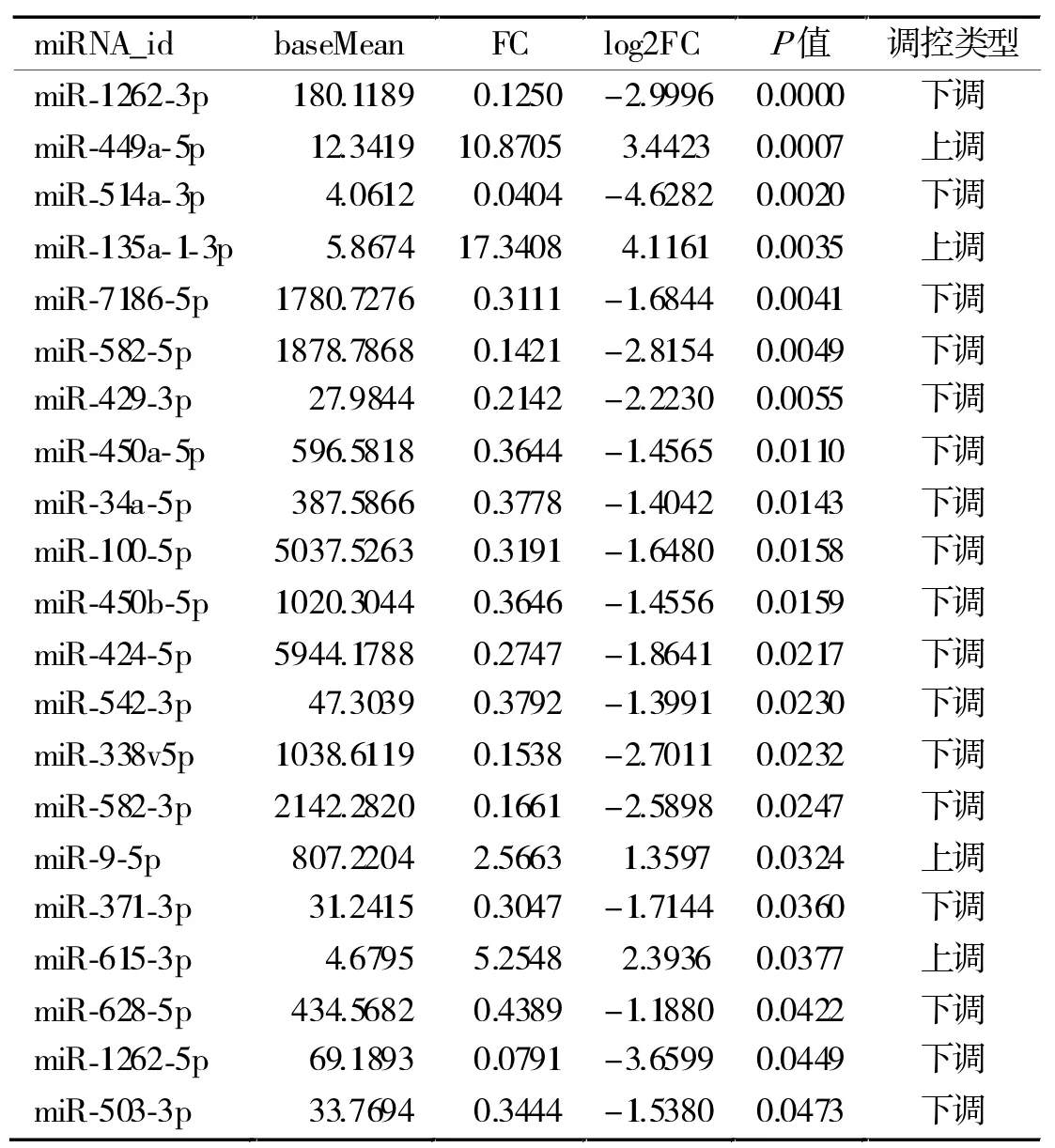
表1 識別的差異表達的已知miRNA
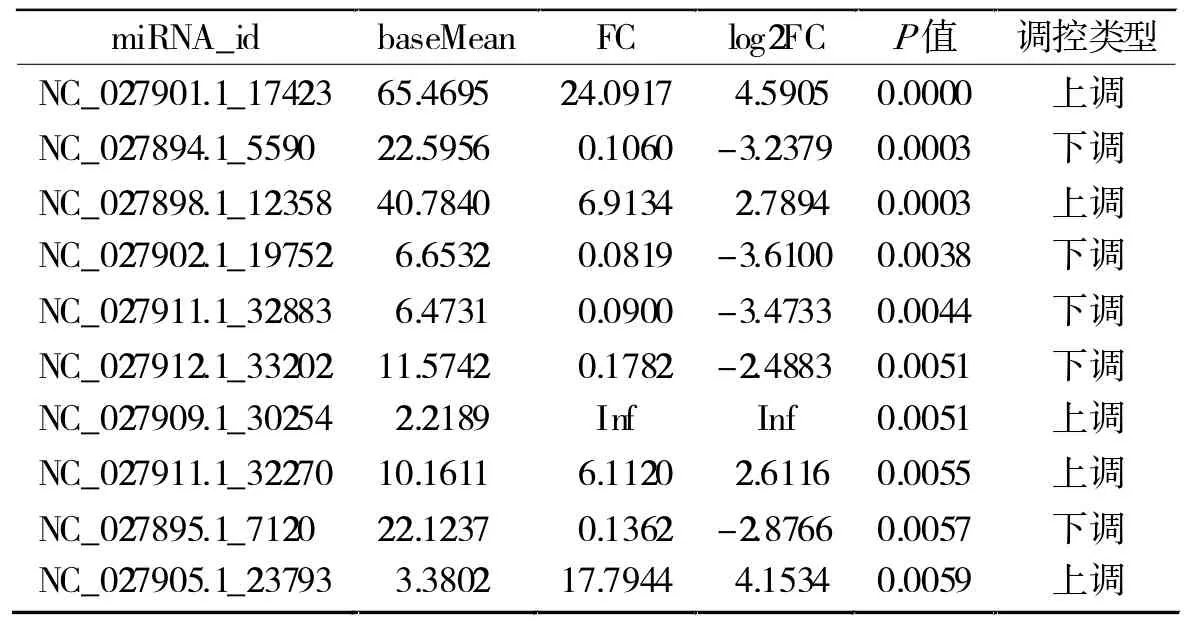
表2 新發現的差異表達miRNA
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國特種設備安全(2018年11期)2019-01-08 02:08:32
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
山東工業技術(2016年15期)2016-12-01 05:31:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:46
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
山東女子學院學報(2014年6期)2014-03-01 02:24:55
