亨廷頓舞蹈病一家系兩例報道并文獻復習
2019-07-12 06:17:06趙紅劉贊華李淑敏藺建文趙紅玲王蘇平
中國全科醫學 2019年20期
關鍵詞:癥狀
趙紅,劉贊華,李淑敏,藺建文,趙紅玲,王蘇平
亨廷頓舞蹈癥(Huntington disease,HD)是一種以神經系統退行性改變為主要特征的常染色體顯性遺傳病,是由4號染色體IT15基因中CAG重復序列異常擴增所致,主要引起紋狀體神經元退行萎縮,疾病癥狀緩慢進行性加重,典型癥狀包括舞蹈樣不自主動作(晚期則運動能力逐漸喪失),精神障礙和進行性認知障礙。HD多發生于中年人,發病年齡40~50歲,但也偶見于兒童和青少年,稱為青少年型亨廷頓癥。世界范圍內的HD發病率約為萬分之一,各種人群均可患病,生存期為10~20年[1]。該病臨床少見,易漏診和誤診。本文對1例臨床疑似HD的家系進行診斷分析,并結合文獻,闡述HD的最新治療進展,以加強對HD的認知。
1 病例簡介
先證者,女,57歲,主因“面部及四肢不自主抽動3年”于2016-12-02就診于大連市中心醫院。患者3年前無誘因出現面部、眼肌、四肢及下頜部肌肉間歇性抽動,幅度小,持續數秒,反復發作,不影響日常生活,患者未察覺被家人發現后,于2013-09-02就診于本院門診,行顱腦CT檢查未見確切異常,未予治療。后患者上述癥狀逐漸加重,不自主抽動發作頻繁,1 min數次,近半年患者自覺記憶力減退,遠期記憶好,近期記憶差,但不影響正常生活,于2016-12-02就診于本院門診,行腦電圖檢查示廣泛輕度異常,為進一步治療收入神經內一科。查體:神清語利,計算力與近記憶力減退,理解力和判斷力正常;眼動充分,未見眼震,雙側瞳孔等大等圓,直徑2.5 mm,對光反射靈敏,雙側鼻唇溝對稱,伸舌居中;四肢肌力正常,四肢肌張力對稱減低,四肢針刺覺對稱存在,面部及四肢間斷細小抽動,雙側指鼻試驗穩準,深感覺對稱正常,雙側Babinski征陰性。輔助檢查:肝功能檢查、腎功能檢查、血糖、血脂檢查、離子、心肌酶、血尿常規均正常;甲狀腺功能檢查正常;腫瘤標志物和自身免疫檢查(抗dsDNA、抗Sm抗體、抗ANA抗體)均正常。心電圖示竇性心律,心電軸左偏,T波改變。顱腦磁共振成像(MRI)示腦內有小缺血性脫髓鞘改變,雙側基底核及皮質萎縮(見圖1);雙側顳葉海馬MRI示皮質萎縮。肝膽脾超聲未見明顯異常;頸動脈超聲未見明顯異常。經顱多普勒(TCD)示顱內外動脈血流未見明顯異常。角膜色素(K-F)環陰性,外周血涂片未見棘紅細胞;電生理示運動神經傳導速度(MNCV)右側脛神經腘窩部波幅降低,感覺神經傳導速度(SNCV)正常,雙側尺神經F波正常,雙側脛神經H反射正常。簡易智能精神檢查量表(MMSE)評分27分,蒙特利爾認知評估量表(MoCA)評分19分(主要表現為執行功能和延遲回憶差)。
家系調查顯示,該家系3代人共12例成員,其中3例發病,1例已故為先證者父親(Ⅰ1)。Ⅰ160歲左右發病,肢體舞蹈樣不自主扭動明顯,疾病后期不能下床行走,后死亡。先證者哥哥(Ⅱ3)55歲左右發病,肢體舞蹈樣不自主扭動,肢體運動不協調,幅度較大,目前使用針灸治療。先證者兒子(Ⅲ1)和先證者哥哥的女兒(Ⅲ2),分別為28歲和30歲,無記憶力下降和肢體扭動。家系系譜圖見圖2。經患者及家屬同意,采集先證者、先證者哥哥和先證者兒子的外周抗凝血5 ml,進行IT15基因檢測,檢測結果顯示,先證者IT15基因CAG重復拷貝數為19/42,確診為HD;先證者哥哥IT15基因CAG重復拷貝數為19/54,確診為HD;先證者兒子IT15基因CAG拷貝數為19/22,未達到基因診斷標準。
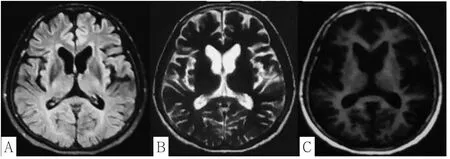
圖1 患者顱腦MRI顯示腦部尾狀核萎縮和側腦室擴大Figure1 The cranial MRI showed atrophy of the caudate nucleus and enlargement of the lateral ventricle
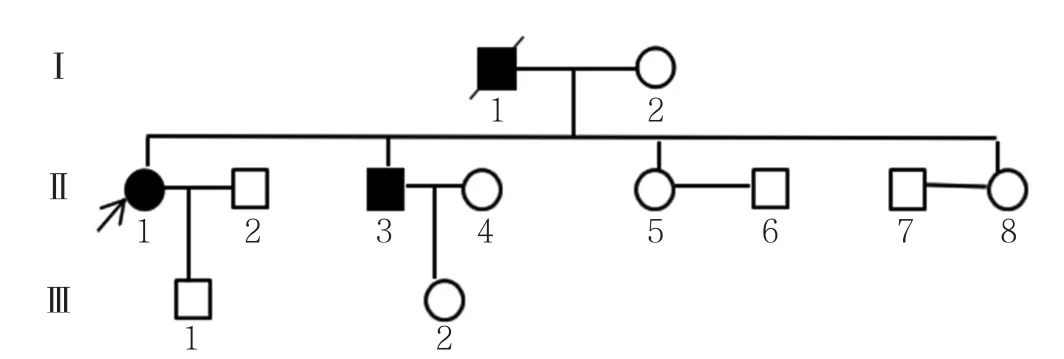
圖2 HD患者的家系系譜圖Figure2 Pedigree chart of HD family
本研究意義:
亨廷頓舞蹈癥(HD)是一類嚴重影響人類健康而缺少任何根本性治療手段的單基因遺傳性神經退行性疾病。自HD致病基因IT15發現以來,近年來科學家們對HD的研究取得了重要突破,揭示了HD產生的遺傳學機制、生化機制、分子機制及腦區特異性機制,將來有望針對這些機制研究出療效更好的藥物,為HD的根本性治療奠定基礎,并對包括HD在內的其他神經變性疾病的治療產生積極的影響。本文對1例臨床疑似HD的家系進行診斷分析,并結合文獻,闡述HD的最新治療進展,以加強對HD的認知。
2 討論
1872年喬治·亨廷頓首次對HD進行了描述,指出其具有遺傳性,并以其名字命名該病[2]。1983年GUSELLA等[3]通過遺傳連鎖分析將HD基因定位于4p16.3區域。1993年,HD協作研究組通過外顯子擴增和cDNA克隆技術,確認CAG重復突變可導致HD發生,并發現了其致病基因IT15[4]。本文對1個家系的發病情況進行了調查,并對家系成員的IT15基因CAG重復拷貝數進行檢測。
2.1 臨床特點 HD是一種常染色體顯性遺傳的神經退行性疾病,由位于4號染色體4p16.3區域的IT15基因CAG三核苷酸重復序列異常擴增所致[4-5]。正常人群中CAG重復序列的長度為6~35個重復,如果CAG重復拷貝數超過40個,則會導致發病,出現異常運動癥狀;如果CAG重復拷貝數為36~39個,部分患者會發病,部分患者會繼續保持無癥狀狀態;CAG重復拷貝數為27~35個時屬于可引起突變的等位基因,個體不會發病;但在減速分裂時不穩定,易發生擴展突變,使其后代患病,尤其是父系遺傳時;CAG重復拷貝數<27個時為正常等位基因,個體正常,不引起疾病。CAG重復拷貝數>36個即為異常,重復拷貝數越高,發病年齡越早,癥狀越重[6]。HD平均發病年齡為40歲,青少年(<20歲)和老年(>70歲)也有發病。病因主要是家族遺傳或者基因受到外部刺激而發生突變,只要雙親任一方遺傳缺陷的基因,皆會表現出病征。HD的病理過程涉及多個方面,包括神經炎性反應、興奮性氨基酸毒性作用、線粒體功能異常、轉錄失調等。病理機制仍未明確,認為CAG過度擴增,機體錯誤的制造一種名為“亨廷頓蛋白”的有害物質,這些異常蛋白質聚集成塊,損害部分神經元,導致患者神經系統退化,并能發展為癡呆[7]。
HD主要累及紋狀體和大腦皮質,具有高度的區域選擇性。基底核運動通路受損引發運動過度—舞蹈樣動作;大腦皮質受損導致患者認知功能障礙。神經影像學檢查作為HD診斷的重要補充參考依據之一,顱腦CT及MRI示基底核及皮質萎縮,萎縮程度可能受CAG重復拷貝數的影響。本研究先證者顱腦MRI顯示腦室、腦池稍大,腦溝稍寬,基底核區萎縮,以尾狀核頭部萎縮最明顯,雙側側腦室額角呈球形向外膨出,側腦室尾狀核區形成特征性的“蝴蝶征”,符合HD特征性影像學表現。本研究的先證者癥狀不典型,其肢體不自主抽動幅度小,不易察覺,沒有明顯的舞蹈樣動作,雖有認知功能減退,未影響日常生活,易漏診,經基因確診為HD,其癥狀不典型考慮與其CAG重復拷貝數少有關。先證者哥哥癥狀典型,肢體舞蹈樣扭動幅度大,認知功能減退明顯,考慮與CAG重復拷貝數多有關。先證者兒子,30歲,無記憶力下降,無精神癥狀,無肢體扭動,基因檢測未達到診斷標準。但有研究報道,1%的HD患者IT15基因檢測顯示陰性[8],故針對先證者兒子應長期隨訪,觀察其癥狀發展。CAG重復序列與臨床表現嚴重程度、影像學改變是否存在正相關,仍需進一步探討。另外,對CAG重復拷貝數的檢測除了對患者確診外,更重要的是對患者及其子女提供咨詢服務,以減少有害突變基因在家族中的遺傳。
2.2 治療與展望 目前仍未發現治療HD的特異性藥物,臨床以對癥治療為主,針對病因、病機的治療目前處于臨床前階段。丁苯那嗪是唯一被美國食品藥品監督管理局(FDA)認可的用于治療HD舞蹈動作的藥物之一,可改善患者不自主運動癥狀,但可能會加重患者抑郁等精神癥狀[9]。抗帕金森藥物可改善運動遲緩和肌強直,常見的有左旋多巴、金剛烷胺等。HD相關認知功能障礙尚無有效藥物治療[10],對于抑郁、焦慮及其他精神癥狀可給予抗抑郁(西酞普蘭等)及抗精神病藥物(奧氮平、利培酮等)改善癥狀。針對病因的治療包括基因療法和分子療法,雖然針對病因的治療難以實現,但針對HD發病機制的不同分子通路正在展開大量的研究,以期減少疾病發生。
線粒體是細胞內的多功能細胞器,不僅參與生物代謝能量產生過程,也能通過與內質網的相互作用調整鈣穩態,產生自由基,參與細胞的死亡進程。線粒體在HD中發揮了重要作用,已有研究發現HD患者的細胞線粒體的形態和結構發生了明顯的變化,這些異常變化可以導致神經元功能紊亂,參與HD的發生[11]。因此線粒體可以作為HD的治療靶標。另有研究認為,HD是常染色體不穩定影響神經發生導致的發育性疾病[12],針對發育階段進行基因調控成為新的治療方法。
HD是由于γ-氨基丁酸(GABA)能神經元受損引起神經環路紊亂,進而導致患者出現運動功能障礙和認知能力喪失等一系列癥狀,GABA能神經元是分泌GABA的重要抑制性神經遞質,對機體的多種功能有重要的調節作用[13]。如果腦內大量的GABA能神經元死亡,就會產生舞蹈樣動作。將GABA能神經元移植到HD患者腦內,從而修復腦損傷環路,將成為HD治療的新的方向。
神經營養因子為神經元提供營養性支持。研究發現,在3-硝基丙酸(3-nitropropionicacid,3-NP)致小鼠HD模型中,神經營養因子3(neurotrophin-3,NT-3)及其受體酪氨酸受體激酶C(tyrosine kinase receptors C,TrkC)mRNA及蛋白表達水平發生改變,NT-3通過TrkC受體調節紋狀體區域的神經傳遞及突觸可塑性[14]。神經營養因子對HD具有神經保護作用,通過遺傳工程使細胞分泌神經營養因子并移植到紋狀體[13],為治療這一疾病帶來了新的希望。
MicroRNA(miRNA)是長度為20~24個核苷酸的內源性小RNA,可以與靶標mRNA的3'UTR結合,調節蛋白質的表達轉錄。由于miRNA片段短,相對分子量小,能夠通過血-腦脊液屏障,且在外周血中有較為穩定的表達,成為多種疾病診斷治療及預后監測的新型生物標志物。近年來發現miRNA可以調節神經退行性相關基因的表達,參與多種神經退行性疾病如阿爾茨海默病、HD、帕金森病等發生[15]。有研究發現HD患者腦脊液中6種miRNA表達增高,且隨著疾病風險增加而升高,這6種miRNA有望作為潛在的生物標志物[16]。以后可以研究如何特異地調節體內miRNA水平,通過降低miRNA的水平,從而推遲疾病的發作。
HD的治療傳統方法很難奏效,雖然基因治療如外源性基因導入[17]、基因沉默[6,18-19]、細胞移植[20]等使 HD 的治療有了新的方向,然而這些方法尚處于探索之中,任重而道遠,期待更多研究為HD的治療帶來新的曙光。有關HD進展的生物標志物的研究正在不斷進行,HD的治療前景仍很樂觀。
作者貢獻:趙紅、劉贊華進行文章的構思與設計,文獻/資料整理;李淑敏、趙紅玲進行文章的可行性分析;藺建文、趙紅玲進行文獻/資料收集;趙紅撰寫論文;李淑敏、藺建文進行論文的修訂;趙紅、王蘇平負責文章的質量控制及審校,對文章整體負責,監督管理。
本文無利益沖突。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
