作為高效水氧化催化劑的多面體Cu2S塊狀晶體溶劑熱合成
2019-08-05 05:39:20魏聰聰鐘達(dá)忠李丹丹郭文君郝根彥
太原理工大學(xué)學(xué)報(bào) 2019年4期
關(guān)鍵詞:催化劑
魏聰聰,鐘達(dá)忠,李丹丹,王 強(qiáng),郭文君,郝根彥,趙 強(qiáng)
(太原理工大學(xué) 精細(xì)化工研究所,氣體能源高效清潔利用山西省重點(diǎn)實(shí)驗(yàn)室,太原 030024)
化石燃料儲(chǔ)備短缺加上環(huán)境污染日益加劇迫切需要開(kāi)發(fā)可再生和更清潔的能源。利用豐富的太陽(yáng)能、風(fēng)能等將其轉(zhuǎn)換為驅(qū)動(dòng)電解水制氫的能量源通過(guò)電解水轉(zhuǎn)換成氫氣進(jìn)行能量的轉(zhuǎn)化、儲(chǔ)存和運(yùn)輸是目前有效的方法之一。電解水制氫過(guò)程包括兩個(gè)半反應(yīng),其中析氧反應(yīng)(OER)通常經(jīng)歷緩慢的4電子轉(zhuǎn)移過(guò)程來(lái)形成O—O鍵,往往因?yàn)閯?dòng)力學(xué)不足,成為電解水過(guò)程的限速步驟。迄今為止在酸性或堿性介質(zhì)中性能優(yōu)異的OER催化劑是基于貴金屬的氧化物,如IrO2和RuO2[1-3].RuO2在堿性條件下具有低至140 mV的過(guò)電位[4]。但是由于貴金屬催化劑昂貴稀缺,在OER方面的應(yīng)用受到限制,因此需要尋找一種低廉、高效、穩(wěn)定性好的催化劑,以此來(lái)降低反應(yīng)的過(guò)電位和電解水過(guò)程的能源消耗,達(dá)到能源的高效利用是關(guān)鍵。
在過(guò)去幾十年里基于非貴金屬Ni,Co,F(xiàn)e基電催化劑在電解水中展現(xiàn)出優(yōu)異的性能。銅作為含量豐富且價(jià)格低廉的金屬和鎳鈷等金屬比較更容易得到氧化態(tài)[5]。但是由于在電解水陽(yáng)極析氧過(guò)程中容易造成陽(yáng)極腐蝕,因此銅基電催化劑在催化水解中的尚未得到很好的研究。為了解決這個(gè)問(wèn)題,有報(bào)道稱氧化銅(CuO)具有富氧化還原性質(zhì),可以作為OER催化劑[6]。DU et al[7]報(bào)道了CuO/Cu foil納米線在小過(guò)電位580 mV下可以達(dá)到10 mA/cm2的電流密度,但是此類催化劑的陽(yáng)極析氧性能還有待提高。HAO et al[8]開(kāi)發(fā)了一種Cu3P/NF納米薄片,其電流密度為10 mA/cm2,過(guò)電壓為290 mV.ZHANG et al[9]以Cu2S為前驅(qū)體經(jīng)過(guò)電化學(xué)線性掃描合成了生長(zhǎng)于泡沫銅(CF)上的三元納米多孔硫摻雜氧化銅Cu2OxS1-x/Cu,表現(xiàn)出優(yōu)異的全水解性能。大量研究表明,引入其他功能材料可以大大提高銅基催化劑的催化性能。XIE et al[10]從CoCO3(OH)2中開(kāi)發(fā)出具有高活性面積的Cu(OH)2@CCHH(CoCO3(OH)2·nH2O)NW/CF核殼納米線陣列,暴露更多活性位點(diǎn),大大增強(qiáng)了OER活性。之前的研究啟發(fā)尋找新的銅基電催化劑,探索提高活性的有效途徑。
金屬硫?qū)倩镒鳛閮?yōu)良的半導(dǎo)體材料,因?yàn)榫哂袃?yōu)良的電導(dǎo)性(快速電荷轉(zhuǎn)移能力)和抗酸/堿性電解液腐蝕的能力,最初應(yīng)用于析氫反應(yīng)(HER),為了挖掘其潛在的雙功能特性,這些材料逐漸在OER方面發(fā)揮作用,傳統(tǒng)的堿性電解液陽(yáng)極析氧催化劑過(guò)渡金屬硫化物(TC,T=Ni,Co,Cu,F(xiàn)e;C=S,Se,Te)在OER中通常表現(xiàn)出較強(qiáng)的催化性能,被報(bào)道的一般在10 mA/cm2的電流密度下,過(guò)電位在200~300 mV的范圍內(nèi)[11-15]。近年來(lái),許多物理和化學(xué)合成技術(shù)廣泛應(yīng)用于合成具有可控形貌、尺寸、組成和結(jié)構(gòu)的新型過(guò)渡金屬硫化物納米材料中,合成過(guò)渡金屬硫化物常用合成方法包括熱注入法、溶劑熱法、混合溶劑熱法、超聲法等。其中,溶劑熱法可較容易的控制催化劑的尺寸和形貌,選擇一種合適的溶劑往往可用作形貌控制器。在混合溶劑熱法中,溶劑往往由兩種及以上組分組成,通過(guò)調(diào)節(jié)組分比例,可以控制催化劑的生長(zhǎng)。當(dāng)有機(jī)胺存在時(shí),就可以很容易地控制產(chǎn)品的成核和生長(zhǎng)過(guò)程,并能協(xié)助晶體各向異性生長(zhǎng)[16]。LIU et al[17]以氯化銅和尿素為前驅(qū)體,在90~110 ℃下一步合成CuS,在胺的調(diào)控下,當(dāng)把三亞乙基二胺(TEDA)和二丁胺(DBA)作為連接媒介時(shí),可以分別合成納米線、納米管和納米囊泡結(jié)構(gòu)。溶劑的pH值、反應(yīng)溫度等因素對(duì)催化劑的結(jié)構(gòu)尺寸也起著至關(guān)重要的作用。混合溶劑熱法具有時(shí)間短,結(jié)晶度高的優(yōu)勢(shì)因此被廣泛的應(yīng)用。
本文以泡沫銅(CF)和硫代乙酰胺(TAA)為銅源和硫源,采用混合溶劑法制備得到結(jié)晶性良好的十四面體Cu2S催化劑。通過(guò)調(diào)節(jié)溶劑乙醇胺與乙醇的比例,較為容易地控制產(chǎn)品的成核和生長(zhǎng)過(guò)程,對(duì)Cu2S結(jié)構(gòu)的完整性和尺寸大小起著至關(guān)重要的作用。Cu2S負(fù)載到泡沫銅上作為陽(yáng)極析氧催化劑在電化學(xué)性能測(cè)試中表現(xiàn)出優(yōu)異的電化學(xué)活性和穩(wěn)定性,并用X射線粉末衍射(XRD),能量分散光譜(EDS),X射線光電子能譜(XPS),對(duì)催化劑進(jìn)行結(jié)構(gòu)、形貌和相組成的表征。
1 實(shí)驗(yàn)
1.1 試劑
泡沫銅(CF,電池級(jí),山西動(dòng)力電池材料有限公司),乙醇胺(ETA,分析純,天津市申泰化學(xué)試劑有限公司),無(wú)水乙醇(ET,分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司),硫代乙酰胺(TAA,分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司),超純水(≥18 MΩ/cm,太原理工大學(xué)煤化工研究所),濃鹽酸(HCl,質(zhì)量分?jǐn)?shù)為36%,永飛化學(xué)試劑有限公司),丙酮(分析純,天津風(fēng)船化學(xué)試劑科技有限公司)。實(shí)驗(yàn)過(guò)程所使用的超純水由GWA-UN1-10C型超純水器制備,其他試劑均為分析純,且在使用前均未進(jìn)一步純化。
1.2 泡沫銅基底的制備
采用泡沫銅(CF)為制備電極的基底。實(shí)驗(yàn)前需要預(yù)處理泡沫銅,首先將大塊的泡沫銅(120 mm×120 mm,厚度0.5 mm)裁剪成實(shí)驗(yàn)條件所需的尺寸(20 mm×30 mm),然后置于丙酮中進(jìn)行10 min超聲清洗,再分別用濃度為2 mmol/L的HCl、乙醇、超純水分別超聲清洗10 min,烘干備用。
1.3 RuO2負(fù)載電極的制備
在0.5 mL(Vethanol∶VH2O=1∶4)的混合溶液中加入1 mg商用RuO2和10 μL質(zhì)量分?jǐn)?shù)5% Nafion溶液,在超聲作用下約1 h形成均勻墨汁狀液體。然后,將墨汁狀液體點(diǎn)涂到CF基底上。催化劑質(zhì)量負(fù)載約1 mg/cm2).在室溫下自然干燥待用。
1.4 多面體Cu2S塊狀晶體的合成
采用溶劑熱的方法在自壓反應(yīng)釜中合成Cu2S,首先將0.24 g的硫代乙酰胺(TAA)溶解在16 mL乙醇胺和乙醇Ven∶Vet=1∶7的溶液中,配置好的溶液攪拌均勻后將其轉(zhuǎn)入反應(yīng)釜中。取干凈的泡沫銅浸沒(méi)到反應(yīng)釜的中央,并密封,將其在180 ℃下反應(yīng)6 h,反應(yīng)結(jié)束后,取出負(fù)載了催化劑的泡沫銅基底,用乙醇和超純水沖洗數(shù)次,在60 ℃烘箱中干燥待用,圖1是實(shí)驗(yàn)的反應(yīng)示意圖。
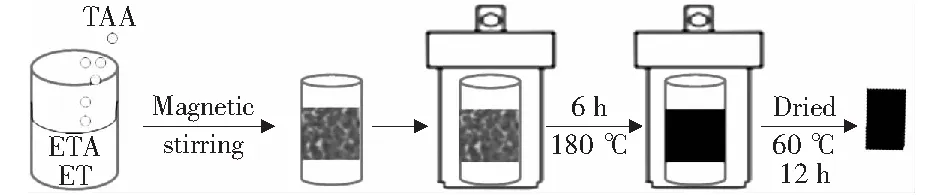
圖1 催化劑合成過(guò)程示意圖Fig.1 Schematic illustration of the synthesis process of catalyst
1.5 樣品的分析和電化學(xué)性能測(cè)試
采用D8 Bruker型X射線粉末衍射儀(X-ray diffraction,XRD)進(jìn)行物相分析。測(cè)試條件為:Cu-Kα (λ=0.154 18 nm),工作電壓40 kV,工作電流40 mA,步長(zhǎng)0.02°,掃描速度8 (°)/min.
掃描電鏡(scanning electron microscopy,SEM)圖譜和X射線能譜(energy-dispersive X-ray,EDS)采用日本Hitachi SU8010型掃描電子顯微鏡和Horiba X-MAX 50能散X射線能譜分析儀測(cè)定。操作電壓:1~3 kV的加速電壓下得到SEM照片;15~25 kV的加速電壓下得到EDS能譜。
利用X射線光電子能譜(X-ray photoelectron spectroscopy,XPS)對(duì)催化劑表面元素的價(jià)態(tài)等進(jìn)行了分析。采用WSCAL-ab220i-XL(VG Scientific,Sussex,UK)型X射線光電子能譜儀進(jìn)行測(cè)定,測(cè)試條件:Al靶,Kα射線,掃描范圍:0~1 000 eV.
本實(shí)驗(yàn)以電化學(xué)工作站(PARSTAT MC,Princeton,USA)為測(cè)試儀器,利用三電極體系來(lái)進(jìn)行測(cè)試,以鉑柱電極為對(duì)電極、Hg/HgO電極為參比電極、Cu2S/CF電極為工作電極。性能測(cè)試環(huán)境條件為常溫常壓,電解液的攪拌速度為50 r/min.電催化析氧測(cè)試是在1 mmol/L KOH溶液中進(jìn)行,在1 mmol/L KOH溶液中進(jìn)行循環(huán)伏安法(CV)測(cè)試,掃描速率為0.05 V/s,電位范圍為0~1 V,直到得到穩(wěn)定的CV曲線。采用線性掃描伏安法(LSV)測(cè)量過(guò)電位,掃描速率為2 mV/s,過(guò)電位公式η=Vapl+pH×0.059-1.23-IR.
2 結(jié)果與討論
以CF和硫代乙酰胺(TAA)分別作為銅源和硫源,乙醇胺(EDA)為絡(luò)合劑,乙醇為溶劑,通過(guò)溶劑熱法制備了負(fù)載于泡沫銅上的多面體Cu2S晶體,并考察反應(yīng)溫度、反應(yīng)時(shí)間、乙醇胺與乙醇的體積比等條件對(duì)催化劑活性的影響。表1分別為實(shí)驗(yàn)的變量和參數(shù),電化學(xué)性能測(cè)試結(jié)果表明(圖2),當(dāng)反應(yīng)溫度為180 ℃,反應(yīng)時(shí)間為6 h,Ven∶Vet=2∶14時(shí)所制備的十四面體Cu2S催化劑性能最佳。
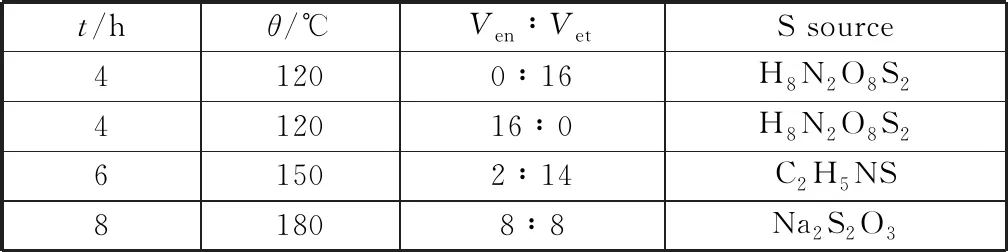
表1 實(shí)驗(yàn)參數(shù)Table 1 Experimental parameters
在醇溶液中乙醇胺(en)發(fā)生水解呈堿性,具有較強(qiáng)的配位能力,可在反應(yīng)體系中激活銅基體表面和電子轉(zhuǎn)移[18-20]。由于胺分子的還原性與銅離子形成配合絡(luò)合物。形成的[Cu(en)2]2+金屬絡(luò)合物與溶液中的 S2-反應(yīng)形成不穩(wěn)定的無(wú)機(jī)-有機(jī)Cu2S-TAA前驅(qū)體,其可作為分子模板控制晶體的生長(zhǎng)[18,21-23]。同時(shí)由于溶液呈堿性,會(huì)形成一部分Cu(OH)2,最后在體系S2-釋放充分的情況下,最終會(huì)得到穩(wěn)定的Cu2S.當(dāng)硫代乙酰胺在上述堿性條件下,發(fā)生水解產(chǎn)生的S2-與銅離子作用反應(yīng)完全時(shí),泡沫銅變?yōu)楹谏罱K得到以泡沫銅為基底負(fù)載有Cu2S的催化劑此反應(yīng)歷程包括兩個(gè)反應(yīng)方程式:
HO(CH2)2NH2+H2O=HO(CH2)2OH+NH3.
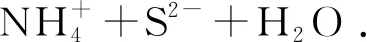
2.1 Ven∶Vet對(duì)Cu2S多面體的形貌和尺寸的影響
當(dāng)反應(yīng)時(shí)間為6 h,反應(yīng)溫度為180 ℃時(shí),取0.24 g的硫代乙酰胺,考察乙醇胺與乙醇的體積比
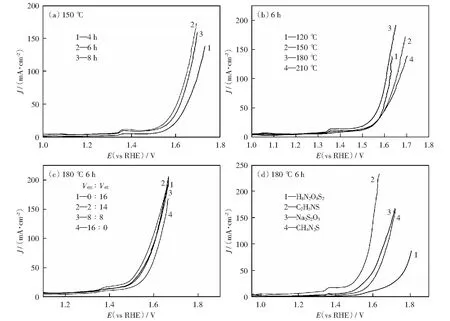
圖2 不同反應(yīng)時(shí)間、反應(yīng)溫度、乙醇胺與乙醇的體積比、S源得到的Cu2S的極化曲線圖Fig.2 LSV curves of Cu2S obtained with different reaction times,temperature,volume ratios of ethanolamine to ethanol,S source
對(duì)電催化性能的影響。乙醇胺在乙醇溶液中水解后顯堿性,相應(yīng)的pH值會(huì)影響溶液中的無(wú)機(jī)鹽和陰離子的水解率(如硫代乙酰胺)[24]。同時(shí)由于有機(jī)胺分子存在[25-26],不僅可調(diào)控催化劑的形貌,也可協(xié)助晶體各向異性生長(zhǎng),并可以嵌入到金屬硫化物(MC)結(jié)構(gòu)中,形成MC-胺-無(wú)機(jī)-有機(jī)絡(luò)合物[27]。溶液只有乙醇沒(méi)有乙二胺時(shí)(如圖3(a)),溶劑極性減小,影響產(chǎn)物的聚集生長(zhǎng)和晶體的取向生長(zhǎng),最終形成不規(guī)則的類球形顆粒。當(dāng)加入乙醇胺時(shí),由于形成了銅胺絡(luò)合物,溶液中自由銅離子濃度較小,體系中產(chǎn)生的Cu2S的速度較為緩慢,有利于各向異性生長(zhǎng);隨著乙醇胺濃度增加體系堿性增強(qiáng),體系內(nèi)的硫源不斷水解,S2-與銅離子快速結(jié)合形成無(wú)規(guī)則Cu2S晶粒,同時(shí)高過(guò)飽和度破壞了晶體的極性生長(zhǎng)環(huán)境,生成大量的無(wú)規(guī)則的Cu2S晶體[23-24,28]。但乙醇胺比例太高時(shí),過(guò)多的乙醇胺又會(huì)阻礙晶體多面體的生長(zhǎng)[29]。當(dāng)Ven∶Vet=2∶14時(shí),晶體具備完整的多面體結(jié)構(gòu),樣品結(jié)晶度較高,催化劑的顆粒尺寸較小、表面積較大,因而Cu2S催化劑的電催化活性較好。

圖3 不同的乙醇胺與乙醇體積比下得到的Cu2S SEM圖Fig.3 Low magnification SEM images of Cu2S obtained with different volume ratios of ethanolamine to ethanol
圖2(c)為不同體積比下的乙醇胺與乙醇所得Cu2S的陽(yáng)極析氧反應(yīng)極化曲線圖。如圖所示,在前面探索的設(shè)定實(shí)驗(yàn)條件下,改變Ven∶Vet的比例,在20 mA/cm2的電流密度下得到不同的過(guò)電位,表2顯示的結(jié)果表明,當(dāng)Ven∶Vet=2∶14時(shí),催化劑表現(xiàn)出最小的過(guò)電位,催化活性最高。
2.2 Cu2S的XRD分析
以硫代乙酰胺為硫源,當(dāng)Ven∶Vet=2∶14,并在180 ℃下反應(yīng)6 h后,得到多面體的Cu2S催化劑,對(duì)此催化劑進(jìn)行物相分析,從XRD圖(圖4)中可以看出催化劑為純相結(jié)構(gòu)的輝銅礦Cu2S,其峰位置與標(biāo)準(zhǔn)卡(JCPDS no.23-961)完全吻合,沒(méi)有其他雜質(zhì)峰出現(xiàn),峰型尖銳、對(duì)稱,表明催化劑的結(jié)晶度較高[30]。
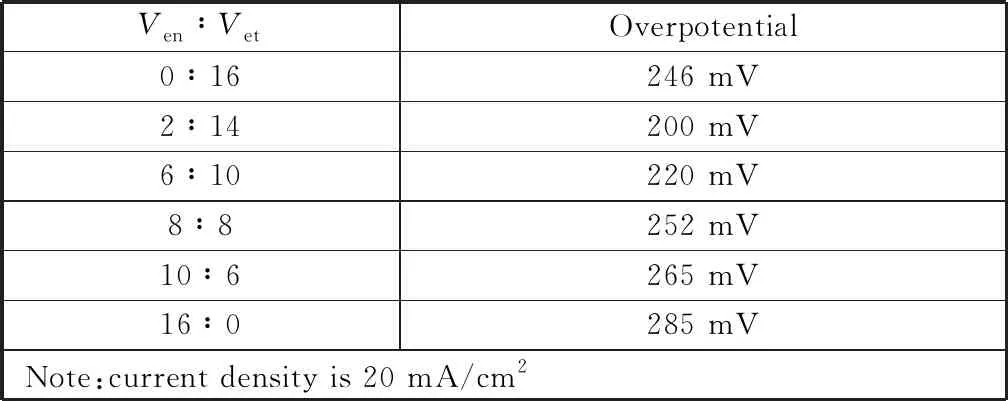
表2 不同體積比的乙醇胺與乙醇下催化劑在1 mmol/L KOH溶液中的OER活性比較Table 2 Comparison of OER catalyst in 1 mmol/L KOH with different volume ratios of ethanolamine to ethanol
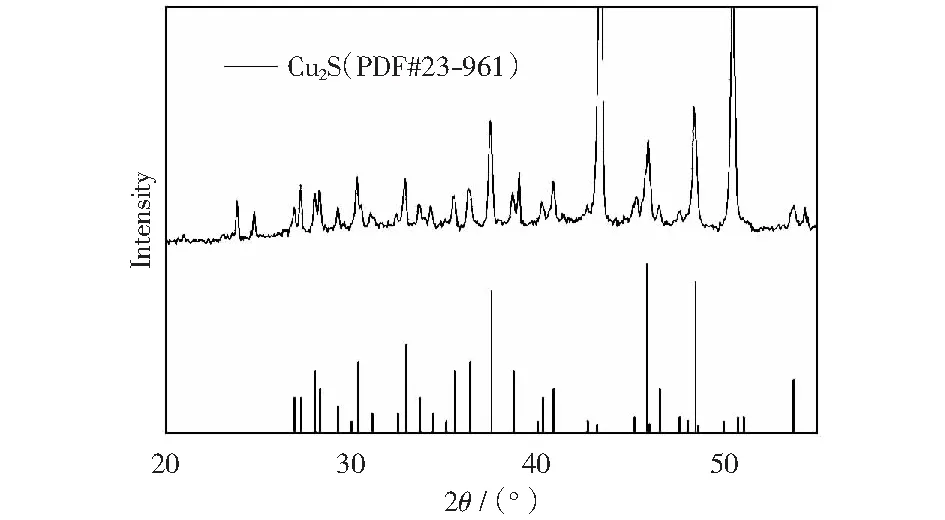
反應(yīng)條件:以硫代乙酰胺為硫源,Ven∶Vet=2∶14,在180 ℃下反應(yīng)6 h圖4 多面體的Cu2S催化劑的XRD圖Fig.4 XRD of Cu2S catalyst was obtained with thioacetamide as the sulfur source
2.3 Cu2S的XPS分析
為了進(jìn)一步確定催化劑的元素價(jià)態(tài),對(duì)催化劑進(jìn)行XPS測(cè)試,分析結(jié)果如圖5所示。Cu 2p的能譜圖顯示:在934.9 eV和955 eV結(jié)合能處的峰位置分別為Cu 2p3/2和Cu 2p1/2,表明Cu2+離子的存在[27];在944 eV和963 eV處為CuO的小衛(wèi)星峰。圖2(a)上Cu 2p軌道結(jié)合能譜,932.85 eV和952.7 eV的兩個(gè)主峰對(duì)應(yīng)的是Cu+2p3/2和Cu+2p1/2的結(jié)合能,與之前報(bào)道的文獻(xiàn)[31-32]一致。二價(jià)銅的出現(xiàn)是由于催化劑表面暴露在空氣中被氧化。對(duì)于圖2(b)S 2p譜,在161.9 eV和 163.1 eV結(jié)合能處顯示了兩個(gè)強(qiáng)峰,證明了S2-的存在即硫化物的形成[32-33],由此進(jìn)一步確定了Cu2S催化劑的形成。
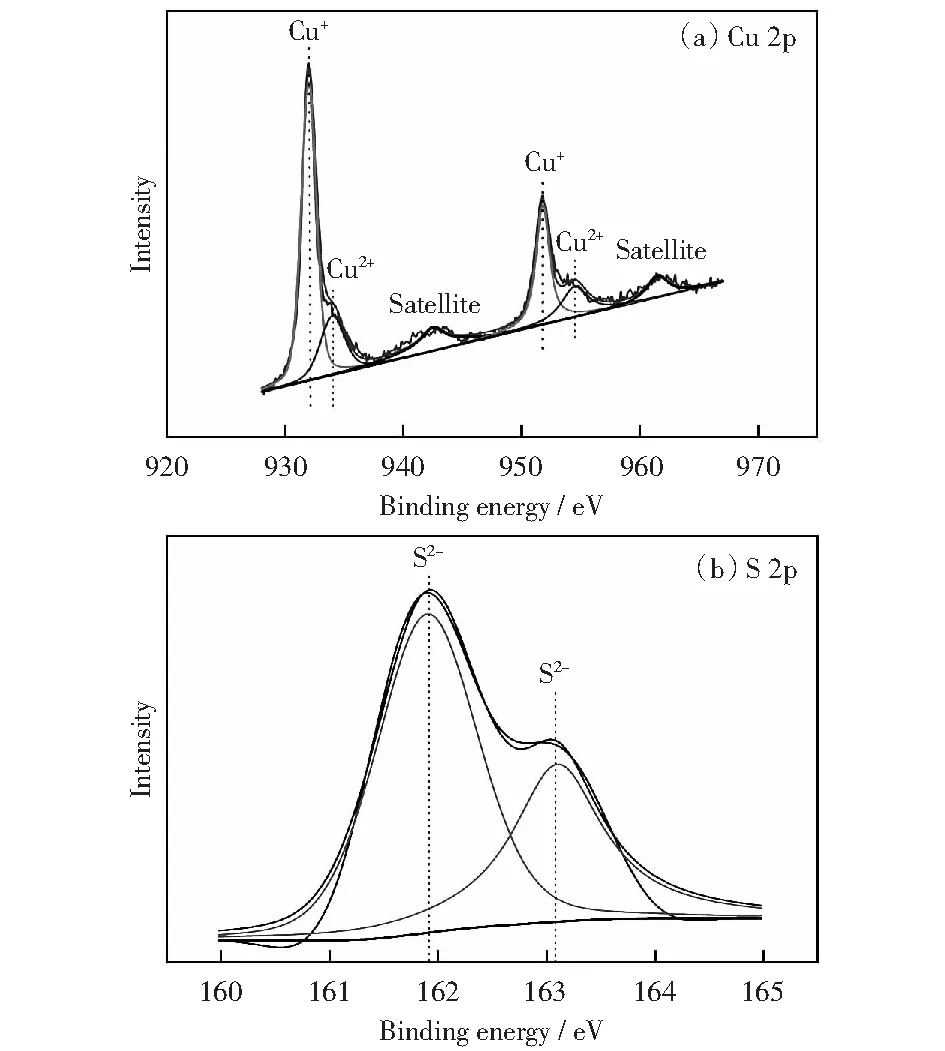
圖5 催化劑Cu2S的XPS譜圖Fig.5 High-resolution XPS spectra
3 Cu2S多面體的陽(yáng)極析氧性能分析
3.1 催化劑的電催化性能測(cè)試
將得到的催化劑在1 mmol/L KOH堿性電解液中進(jìn)行電化學(xué)性能測(cè)試,催化劑在經(jīng)過(guò)多圈CV后被激活,然后采用線性掃描伏安法(LSV)測(cè)試催化劑的電化學(xué)性能。在同樣的體系下分別對(duì)空白的泡沫銅利用滴涂法制備的RuO2/CF和在最佳實(shí)驗(yàn)條件下得到的Cu2S多面體催化劑進(jìn)行電化學(xué)性能的比較。如圖6(a)為三者的LSV曲線,可以明顯的觀察到,在電流密度為20 mA/cm2時(shí),Cu2S 催化劑的過(guò)電位只有200 mV,Cu2S催化劑具有優(yōu)于貴金屬氧化物RuO2(過(guò)電位為306 mV)的OER性能,對(duì)比已報(bào)道的過(guò)渡金屬陽(yáng)極析氧催化劑表現(xiàn)出較好的電化學(xué)性能(表3是近年來(lái)所報(bào)道的陽(yáng)極析氧催化劑的性能對(duì)比)[2,6,8,13,34-39],并且催化劑的穩(wěn)定性是衡量其電化學(xué)性能的重要因素,在不同的外加電壓下,分別得到Cu2S在10 mA/cm2和30 mA/cm2的電流密度下的計(jì)時(shí)電流曲線如圖6(b)所示,催化劑可以保持至少30 h的穩(wěn)定性。
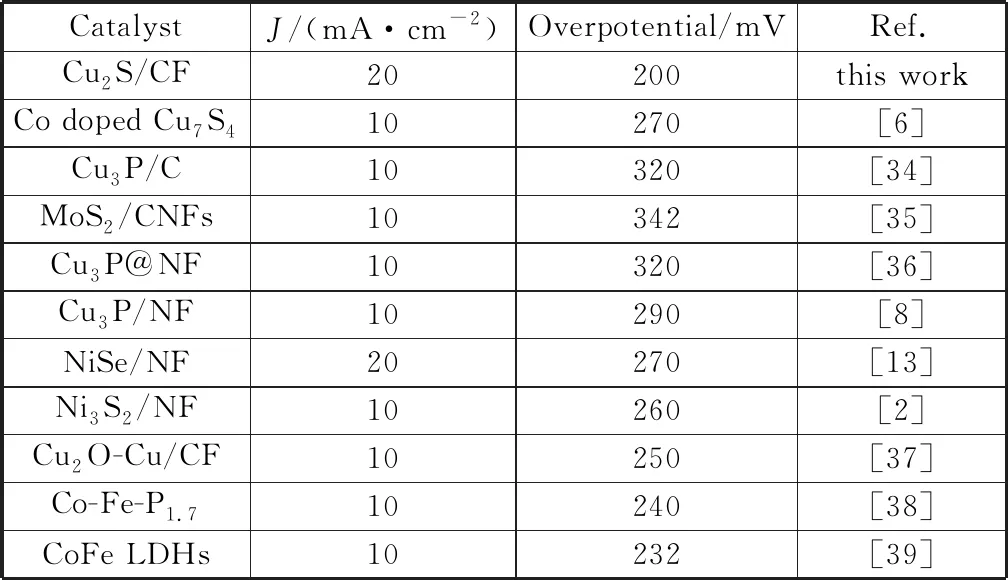
表3 報(bào)道的OER催化劑在1 mmol/L KOH溶液中的活性比較Table 3 Report on the activity comparison of OER catalyst in 1 mmol/L KOH
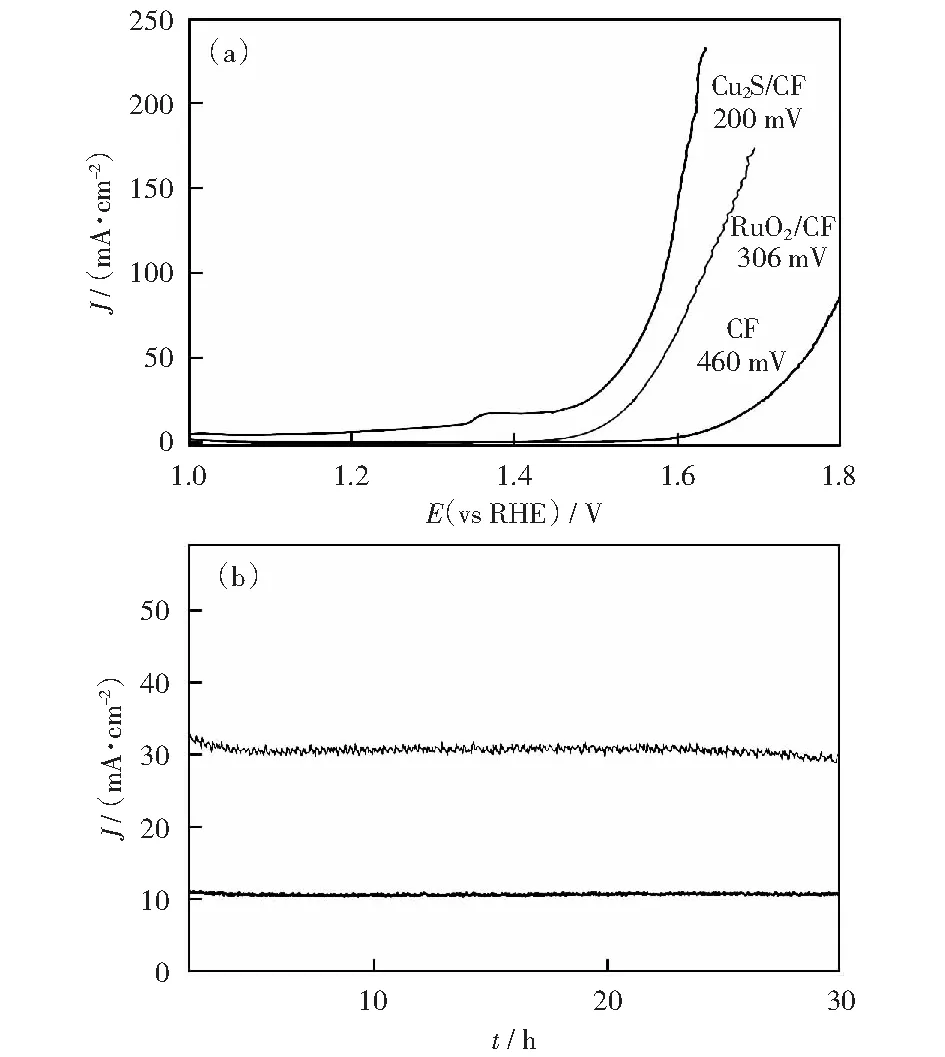
(a) 1 mmol/L KOH中空白CF,RuO2/CF和Cu2S/CF的極化曲線,掃描速率為2 mV/s;(b) Cu2S/CF在10 mA/cm2和30 mA/cm2恒電流密度下的長(zhǎng)期穩(wěn)定性試驗(yàn)圖6 催化劑的電化學(xué)性能Fig.6 Electrochemical performance of the catalysts
3.2 Cu2S多面體催化劑OER測(cè)試前后元素組成與形貌分析
電解水OER催化劑開(kāi)始于金屬氧化物,氧化物是OER中真正起催化作用的物質(zhì),后來(lái)發(fā)展的過(guò)渡金屬硫化物最開(kāi)始應(yīng)用于HER.研究者發(fā)現(xiàn)其在OER過(guò)程中部分或完全轉(zhuǎn)化為氧化物,比過(guò)渡金屬氧化物具有更好的陽(yáng)極析氧性能。XU et al[40]在2016年合成了鎳鐵二硒化物(NixFe1-xSe2),它原位轉(zhuǎn)化為氧化物時(shí),在10 mA/cm2的電流密度下的過(guò)電位僅有195 mV.這種NixFe1-xSe2衍生催化劑的高活性主要是由于其理想的納米結(jié)構(gòu)。
對(duì)多面體Cu2S催化劑OER前后的物質(zhì)組成和晶體結(jié)構(gòu)進(jìn)行分析,探究其電化學(xué)陽(yáng)極析氧過(guò)程。通過(guò)EDS能譜圖和元素映射圖(見(jiàn)圖7),考察催化劑在OER前后元素及其含量的變化,通過(guò)對(duì)比發(fā)現(xiàn),S元素存在流失現(xiàn)象,同時(shí)O元素有明顯增多。對(duì)于OER測(cè)試后的物質(zhì)組成與元素分析通過(guò)XPS分析(見(jiàn)圖8).S 2p的峰在OER測(cè)試之后幾乎消失,O 1s譜圖中顯示的主峰值531.6 eV,代表Cu—O;530.2 eV的峰歸因于CuO的晶格氧[33]。Cu 2p的峰由原來(lái)代表Cu2S中兩對(duì)2p3/2Cu+和2p1/2Cu2+的峰變?yōu)榱薈uO的峰,峰值出現(xiàn)在933.6 eV和 953.6 eV的結(jié)合能處其歸因于Cu 2p3/2和Cu 2p1/2,它是CuO形式的Cu2+.同時(shí)XRD分析(圖8(d))也表明Cu2S相消失,CuO相形成。
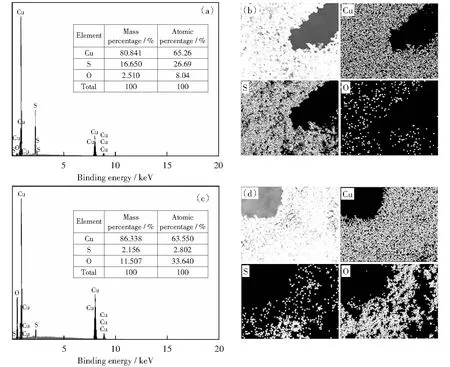
圖7 OER前后催化劑的EDS譜圖和對(duì)應(yīng)的元素的映射圖像Fig.7 EDS and corresponding Elemental mapping images before and after OER. The inset in the spectra shows the elemental atomic percentages.(a,b) the as prepared Cu2S/CF,(c, d) Cu2S/CF after OER
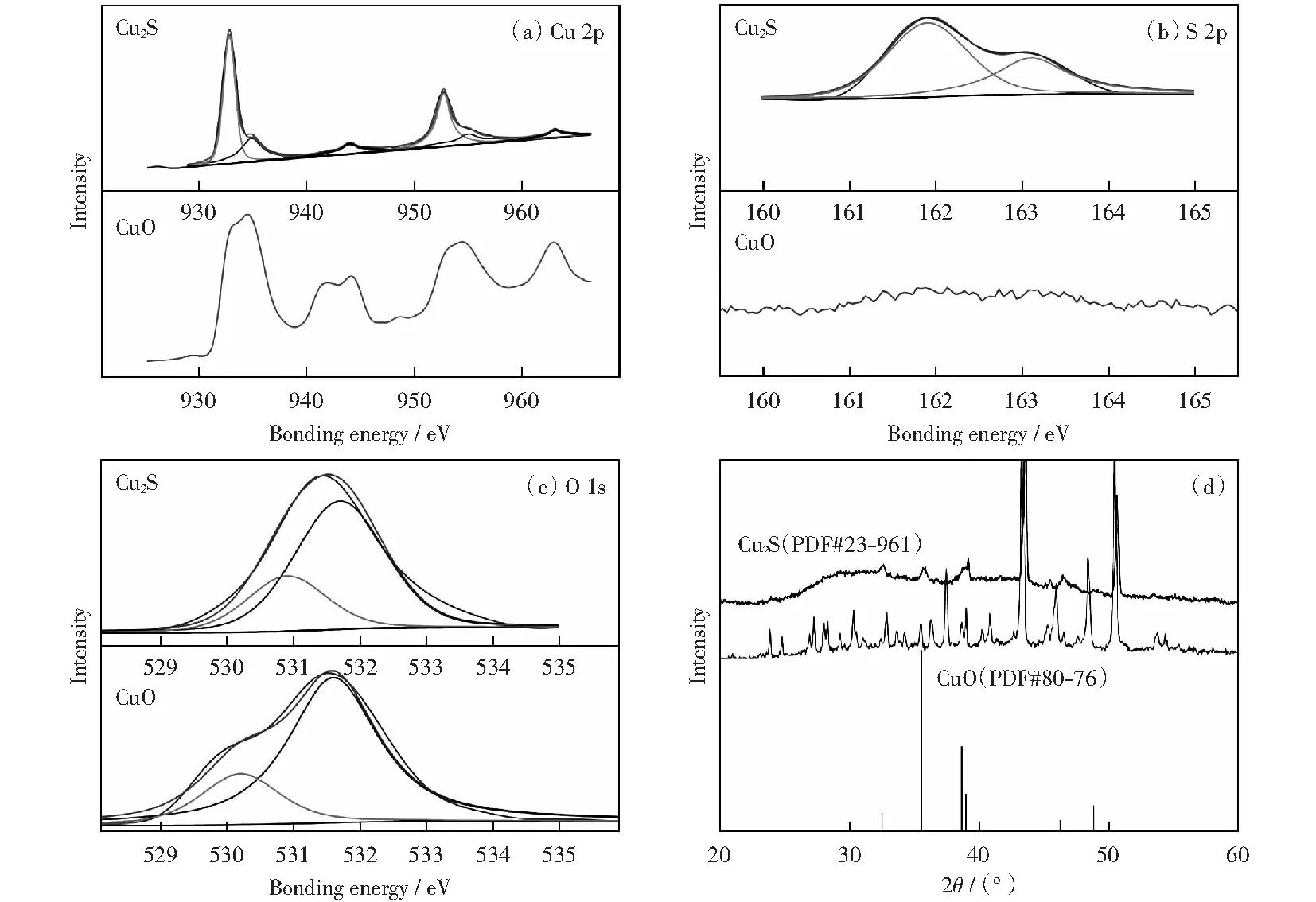
圖8 Cu2S OER反應(yīng)前后比較Cu 2p,S 2p,O 1s的XPS譜及XRD圖Fig.8 Comparison of Cu2S before and after OER, High-resolution XPS spectra of Cu 2p, S 2p, O 1s and XRD patterns

圖9 負(fù)載于泡沫銅上的Cu2S催化劑OER前后的SEM圖形貌Fig.9 SEM images of structual characterization of catalysts Cu2S loaded on copper foam before and after OER
對(duì)催化劑在1 mmol/L KOH 中OER測(cè)試前后的形貌進(jìn)行了對(duì)比,測(cè)試前的Cu2S催化劑為由兩個(gè)六邊形的面和六對(duì)梯形面組成的十四面體形貌(如圖9(a)(b)).晶塊的尺寸以上下2個(gè)六邊形的兩點(diǎn)最長(zhǎng)距離為基準(zhǔn),平均晶體直徑大約為6 μm.在對(duì)催化劑進(jìn)行活化時(shí),更有利于其轉(zhuǎn)化為活性物質(zhì)。通過(guò)Cu2S OER測(cè)試前后形貌的對(duì)比發(fā)現(xiàn),Cu2S十四面體變的粗糙,經(jīng)過(guò)電化學(xué)氧化過(guò)程出現(xiàn)分層,像是花團(tuán)錦簇的花環(huán),催化劑的比表面積進(jìn)一步增大見(jiàn)圖9(c)(d).顯然活化后的催化劑具有更優(yōu)的形貌,暴露更多的活性位點(diǎn),電荷轉(zhuǎn)移速率加快,促進(jìn)了電解水陽(yáng)極析氧反應(yīng)。
4 結(jié)論
本實(shí)驗(yàn)主要通過(guò)液相合成法通過(guò)調(diào)節(jié)反應(yīng)體系中各個(gè)反應(yīng)參數(shù)探索反應(yīng)溫度、反應(yīng)時(shí)間、乙醇胺與乙醇的體積比等對(duì)催化劑活性的影響,最終得到以硫代乙酰胺為硫源,當(dāng)Ven∶Vet=2∶14,在180 ℃下反應(yīng)6 h,得到十四面體的Cu2S催化劑具有最好的析氧活性。在探索過(guò)程中得到以下結(jié)論:
1) 在電流密度為20 mA/cm2時(shí),Cu2S催化劑的過(guò)電位只有200 mV,對(duì)比已報(bào)道過(guò)的過(guò)渡金屬陽(yáng)極析氧催化劑表現(xiàn)出更優(yōu)的電化學(xué)性能。
2) 調(diào)節(jié)乙醇胺與乙醇的比例,對(duì)Cu2S結(jié)構(gòu)的完整性和尺寸大小起至關(guān)重要的作用。
3) 反應(yīng)的溫度和溶液的pH值會(huì)影響無(wú)機(jī)鹽和陰離子的水解率(如硫脲和硫代乙酰胺),從而影響其晶體結(jié)構(gòu)中固有的各向異性特征,導(dǎo)致晶體的定向生長(zhǎng)。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時(shí)代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國(guó)資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
