美國藥品上市后再評價法律制度實施的研究及其對我國的啟示Δ
2019-08-15 02:11:56張琪顏建周馬旭鋒邵蓉
中國藥房 2019年15期
張琪,顏建周,馬旭鋒,邵蓉
(中國藥科大學國家藥物政策與醫藥產業經濟研究中心,南京211198)
藥品上市后再評價(以下簡稱“再評價”)指從藥理學、藥劑學、藥物經濟學等方面對已批準上市的藥品在社會人群中的療效、不良反應、費用及是否安全、有效、經濟合理所進行的科學評價。美國是最早開始進行再評價的國家之一,并且目前已經構建了相對比較成熟的再評價法律制度體系,其實施流程也富有特點。在美國再評價體系下,FDA對上市后藥品的安全風險進行實時監測,以期有效控制風險[1]。
2015-2018 年,我國在藥品審評階段實施了一系列改革措施(如《國務院關于改革藥品醫療器械審評審批制度的意見(2015)》[2]、《關于深化審評審批制度改革鼓勵藥品醫療器械創新的意見(2017)》[3]、《關于鼓勵藥品創新實行優先審評審批的意見(2017)》[4]),藥品上市速度得以加快,但為了在審評加快的背景下保證品種安全,需要良好的上市后再評價體系的支撐。目前我國的再評價體系存在再評價方法不明確、缺乏企業舉證責任等問題[5]。本文將對美國的再評價體系進行梳理,重點研究美國再評價法律制度實施現狀與程序,為我國建立并完善再評價法律制度提供借鑒[6]。
1 美國再評價法律制度的實施背景及發展歷程
1.1 美國再評價法律制度的提出背景
美國以1962年“反應停”事件為背景,提出不僅應檢查藥品安全性也要跟蹤上市后藥品有效性的觀點[7];1984年《藥品價格競爭與專利期補償法案》(Drug Price Competition Patent Term Restoration Act)的頒布使大量仿制藥上市,但1989年仿制藥欺詐丑聞令美國推行大范圍一致性評價工作。
1.2 美國再評價制度的具體發展歷程
美國再評價體系自20世紀60年代開始建設,通過頒布一系列法案和開展大范圍再評價活動,逐步成熟并成為全球標桿。
1.2.1 再評價工作初創階段 1962年,《聯邦食品、藥品和化妝品法案》(Fedral Food,Drug&Cosmetic Act)正式對藥品上市后安全性檢測作出規定,要求藥品企業應當定期向FDA報告產品的不良反應情況并同時規定在FDA可以確定藥品的不良反應達到危及公眾健康的程度時有權進行相關藥品的撤市工作[8]。
“反應停”事件迫使美國FDA于1962年修改1938年的藥品法律法規,美國國立科學院(National Academy of Sciences)成立國家科學研究委員會(National Research Council),對1962年以前批準的所有藥品的功效、價格等進行統計調查,并建立執行藥效研究實施方案(Drug efficacy study implementation,DESI)。共審核4 000多份藥品文件,淘汰了大約300種不合格藥品。檢查結果分為了三類:①對其說明書所列適應證均確切有效;②對所列適應證療效不明確;③對所列適應證無效,有2 000多種藥品被認為“確切有效”,760種被認為是“無效的制劑”,約600種藥品被禁止繼續銷售,而剩余的藥品被認為“療效不明確”。自此,美國對藥品風險規管由被動的事后處罰轉向主動的事前控制,由單純的注重安全性轉向安全性與有效性并重[9]。
1.2.2 再評價工作發展階段 1984年,美國頒布《藥品價格競爭與專利期補償法案》,大量仿制藥上市,但不斷出現仿制藥欺詐與質量問題。有學者指出[10],直至1984年仍有607種已被確認缺少有效性證據的藥品在流通。隨后,美國進行第二次大規模探進式再評價工作,對仿制藥進行大范圍的一致性評價。
FDA動用17個檢測中心,300多名專業人員對市場30%的藥品進行分析測試,對于24種治療窗窄(有效劑量和有毒副反應劑量接近)的藥品,FDA進行了至少3批生產批次的抽樣調查[11]。在此過程中,美國FDA主要檢查以下內容:①仿制藥與參比試劑的藥學等效性;②仿制藥與參比試劑的生物等效性;③仿制藥與參比試劑是否具有相同標簽內容;④仿制藥與參比試劑是否具有相同化學、生產和控制相關信息。通過對以上信息核查,給予藥品“A/B”評級,等效藥品將會出現在《通過等效性評估獲得批準的藥物名單》(又名“橙皮書”)當中[12]。
截至1989年底,FDA已分析仿制藥2 500多種,并同時公布生物等效性評價合格目錄,編撰《經治療等同性評價批準的藥品》(Approved Drug Products with Therapeutic Equivalence Evaluation)。FDA從30種處方量最大的仿制藥中抽查2 550種仿制藥樣品和成品,逐一進行樣品質量分析測試;對36家仿制藥企業和12家外包生產廠家進行現場考核和注冊材料抽查。
此次大規模評價工作令美國進一步細化再評價相關文件,1992-2001年相繼出臺一系列有關指南,如《上市后藥品不良事件報告指南》(1992)、《人用藥品和生物制品上市后不良事件報告》(1997)、《人用藥品和生物制品上市后不良事件報告》(2001)等。
1.2.3 再評價工作成熟階段 步入21世紀以來,經過前兩次大規模檢查,美國藥品監管政策改革的重要趨勢是加強上市后的安全監管。2007年的《食品藥品監督管理局2007修正法案》(Food and Drug Administration Amendments Act,FDAAA)提出了如下的要求[7]。
①在行政權力方面,FDA可對上市后的藥品進行研究;并且有權利限制危險系數高的藥品上市銷售;同時可以根據其風險程度的高低,要求藥品生產公司在30 d內對該藥的標簽說明書進行修改。
②在財政資源方面,美國國會將在5年之內批準向FDA撥款2.25億美元專用于FDA專職負責藥品上市后風險管理的監測與流行病學辦公室,加強藥品安全跟蹤監測。
③提升藥品上市安全監管部門的地位和級別,將流行病學辦公室(Office of Surveillance and Epidemiology,OSE)從新藥辦公室中獨立出來。
④宣布對全美國的藥品不良反應報告系統進行大規模更新,并將政府內部大型的藥品不良反應信息網絡與私人企業數據庫進行對接,以實現信息和數據共享。
1996年,美國藥品審評中心(CDER)提出評價質量管理規范(Good review practices,GRP)的概念,制定了評價的指導準則,用來指導再評價的程序,隨后CDER于2007年對GRP進行了完善[7]。
2012年美國又出臺《FDA安全與創新法案》(Food and Drug Administrition Safety and Innovation Act,FDASIA),此次法案是對《聯邦食品藥品化妝品法案》的又一次重要修訂[7]。其在再評價方面的主要進步體現在:①設置唯一設施識別系統(Unique facility identifier system)。該系統用于再評價過程中的藥品追溯,方便修改標簽、撤市、召回,并有利于FDA和企業追溯藥品質量,是藥品唯一編號,也方便FDA使用該系統中的信息對企業進行基于風險的檢查和再評價。②FDA獲藥品行政扣押權。若在再評價過程中或是在檢查過程中認為某藥品摻假或偽標,FDA有權立即簽發扣押令,對藥品實施暫時性扣押,扣押期一般不超過20 d。被扣押的藥品單位需至少提交2年的材料供FDA檢查,哪個環節可能摻假或偽標,這也成為再評價中的重要環節[13]。
經過2007年與2012年相關法案的修改完善,美國再評價體系逐漸完善,主要包括上市后監督、年度報告、風險評估戰略等幾方面內容。發展成熟的美國再評價體系見圖1。
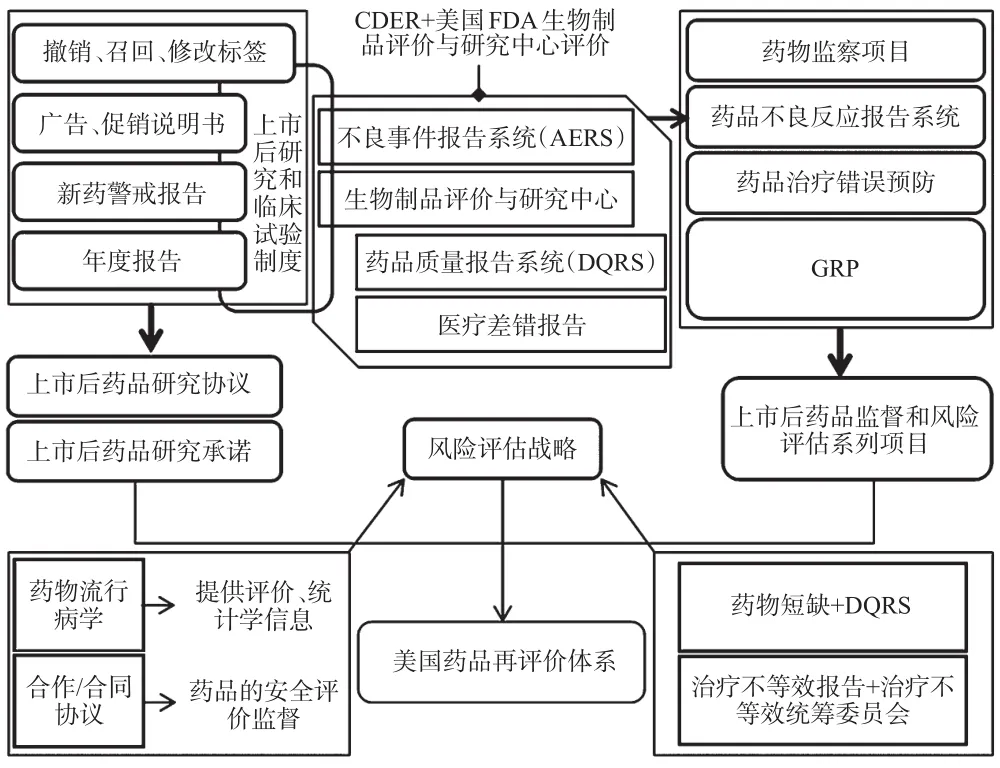
圖1 發展成熟的美國再評價體系Fig 1 American developed drug re-evaluation system
至此,美國形成了以企業為執行主體、政府為監督主體,持續性強、有針對性、發揮社會力量協同治理,過程高度信息化和透明化的再評價體系。
2 美國再評價法律制度的實施現狀
2.1 美國再評價的實施形式
美國再評價工作現今可分為三個部分,分別為藥品不良反應報告和監測制度的實施、定期報告制度的落實與上市后臨床試驗及研究制度的開展。其中,上市后臨床試驗及研究制度又包括上市后研究要求和上市后研究承諾兩部分。
在進行再評價工作時,若發現藥品存在嚴重危及公眾健康的不良反應事件或查處明顯違反相關規定的藥品包裝、質量等問題,FDA將與企業進行商議,主要以企業為主、FDA為輔進行相關藥品修改標簽、召回、撤市等工作。在進行修改標簽、召回、撤市之前的檢查工作表面上由CDER負責(但CDER不檢查藥品),但企業有責任在市場上調查并向CDER遞交證明藥品安全有效的數據。
2.2 再評價制度的實施流程
美國現今對再評價結束后結果處理主要分為修改藥品標簽、召回、撤市3種,其中以修改標簽為主,其次是召回,撤市的藥品非常少。3種情況對應的檢查流程非常相似,通常可以分為發現線索、FDA初步審查并通知、企業深入自查與審查、企業行動并接受FDA監督[14]。
2.2.1 發現線索 藥品上市后,通過匯總新藥警戒報告、年度報告、藥品不良反應報告,進行藥品常規檢查,開展FDA專項行動等途徑,或在企業日常檢查評級過程中,或是接到舉報電話或突發事件通知后,FDA便啟動相應的再評價后續流程,對藥品情況進行進一步檢查和評估[15]。
2.2.2 FDA初步審查并通知 再評價啟動后,通過上述途徑對藥品進行檢查或接收相關信息發現藥品不良狀況后,FDA進行初步審查,對相關信息進行整合,告知責任企業詳細信息并建議其進行自主核查和深入監測,對自身產品實施客觀檢查和評估并做進一步的補救措施[16]。
2.2.3 企業深入自查與審查 在收到FDA通知并與其充分溝通后,大部分企業會接受FDA的建議,展開對問題藥品的檢查,檢查其原料供應、生產環境、生產過程、包裝環境及過程、儲藏運輸條件和標簽說明書是否規范等情況,比照患者的報告或事件的細節在必要時進行臨床/非臨床試驗,統計結果并得出結論,通過嚴謹的評估確定下一步措施[8]。
2.2.4 企業行動并接受FDA監督 在評估結束后,企業一般會采取藥品召回、撤市或修改標簽等措施補救,執行主體主要為企業,FDA起監督作用。召回、撤市、修改標簽主要通過美國藥品唯一設施識別系統向醫療機構、藥店等銷售、使用主體發出通告,實施相關行動,并警示其不要再繼續出售或使用相關藥品[14]。
結合上述內容,美國再評價流程見圖2。
下文將對藥品召回進行進一步說明,分析美國再評價工作的實施流程和詳細操作方法。藥品召回主要指經FDA通知或建議,企業自愿或由FDA強制實施將某藥品從市場上撤出的行動,可能是企業主動行動措施,也可能是在FDA建議下做出,還有可能是FDA依法強制企業實施。藥品召回分為三種不同的等級,分別為一級召回、二級召回和三級召回。典型案例的再評價流程見圖3。
①發現線索。藥品再檢查行動一般由FDA發起,企業主動進行自檢后發現問題的占少數。FDA常通過以下幾種方式獲知藥品的安全性存在隱患,之后對企業提出相關建議。a.進行常規性的再檢查。主要為FDA按《良好藥品臨床試驗規范》(Good Clinical Practice,GCP)、《藥物非臨床研究質量管理規范》(Good Laboratory Practice,GLP)、《良好生產規范》(Good Manufacturing Practice,GMP)進行上市后日常性的檢查工作,如在平日抽檢中,FDA可能會經過采樣測試、分析檢驗后發現藥品的安全性隱患,例如標簽不清晰、注射液存在可見顆粒物質、含未申報的活性藥物成分等。b.FDA收到客戶投訴。FDA會通過Medwatch自愿報告系統Medwatch等通道或是郵信等方式收到消費者、醫護工作者、經銷商的投訴或報告。一般藥品購買者會將遇到的問題報告給FDA,請求FDA責令相關企業召回或撤市。投訴包括了藥害事件發生,突發情況下相關主體提出的訴訟和舉報。Medwatch是美國藥品上市后的不良反應監督計劃,全稱為“安全信息和不良事件報告計劃”(Safety information and adverse event reporting program),適用于醫療衛生專家和消費者。醫務人員、消費者和患者在獲知藥品不良事件后,可直接向FDA報告,也可向生產企業報告。對于消費者與患者,FDA鼓勵其在發生不良反應時先報告給醫師,再由醫師提交報告;如果醫師選擇不報告,患者也可以自己提交。Medwatch平均每年接到大約20 000份不良反應報告。從報告來源看,藥師(32%)報告最多,其次是醫師、護士以及消費者。消費者上報的比例相對較小,但對于所有消費者主動上報的行為,FDA都會以書面信函方式表示感謝[15]。c.FDA收到不良反應報告。FDA會經常性、規律性地查看藥品的不良反應報告,并通過數據分析進行統計和評估。FDA可察覺企業生產的藥品是否存在安全隱患。報告中報告者會簡述相關不良反應癥狀、不良反應出現的時間節點以及用藥的相關情況,由此可推斷出藥品可能出現的安全狀況,方便FDA對藥品生產企業的調查與追詢。線索的發現主體一般為FDA,可以通過常規性再檢查、客戶投訴、不良反應報告這3種渠道獲取藥品存在安全隱患的訊息。少數線索由企業發現,有留樣自查和收到投訴/報告兩種形式(詳見后文)。
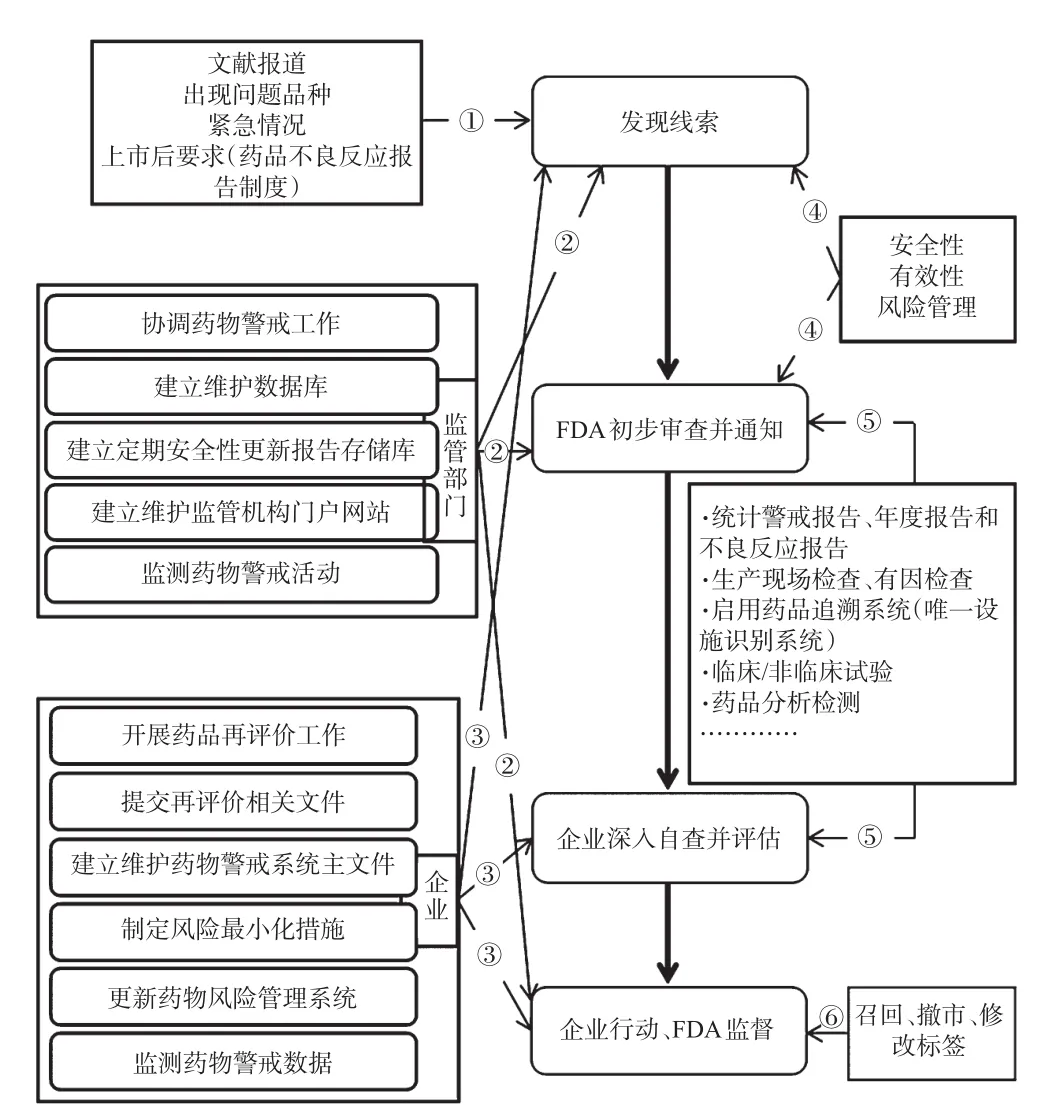
圖2 美國再評價流程Fig 2 American drug re-evaluation process
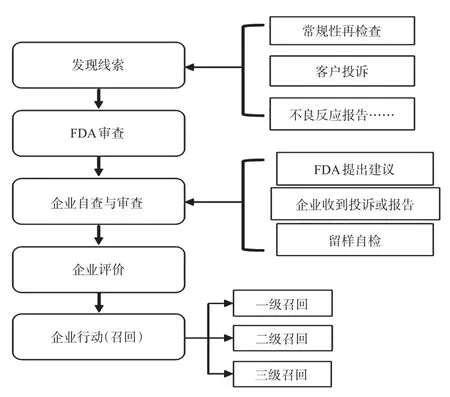
圖3 典型案例的再評價流程Fig 3 Typical cases of drug re-evaluation process
②FDA審查。FDA發現問題會通知企業。FDA將具體情況告知企業,企業自發進行檢查工作,檢查完畢后,企業評估分析結果做出如召回、撤市或修改標簽等行動。所以,發現問題后由FDA進行檢查工作的情形相對較少,如果審查由FDA主導,FDA將到企業現場進行抽樣和相關檢查[16]。
③企業自查與審查。企業自查的起因一般有3個。a.FDA提出相關建議。企業在FDA提出建議或告知相關狀況后會進行自我檢查,查明出現的原因,并通過評估分析,得出解決方案。b.企業收到相關投訴/報告。消費者、醫護工作者或經銷商直接向企業投訴或出具相關報告,企業接到投訴后,立即展開檢查工作。首先確定投訴/報告內容真實性,確定后采取進一步措施進行檢查、溯源、分析和評估出現問題的原因,方便進一步的探究和解決[17]。c.企業自檢(留樣檢查)。主要針對GMP檢查,當政府完成抽樣時,企業至少留樣兩批,在企業存留期間也要對其進行實時查驗,一旦發生問題可隨時應對出具解決方案[18]。在對留樣進行觀察和查驗的過程中,企業可能發現相關安全隱患,進而引發后續的再檢查活動,利用精密儀器、檢查設備,或查看企業生產的原始記錄對藥品進行詳細核查,確定癥結所在并進行整改或召回。
3 美國經驗對我國的啟示
3.1 凸現患者主體作用,引導公眾實現風險溝通
自發報告是在全世界范圍內廣泛應用的藥物警戒辦法。患者作為直接使用者能最快發現并最真實描述藥品不良反應,讓患者主動參與藥品不良反應監測過程,對藥物的風險-效益評估有重要作用[19]。
我國不良反應報告主要來源于醫療機構,其次為藥品生產經營企業,而個人報告總數未達到1%。此外,由于醫療機構存在制度不執行、培訓不嚴格、人員工作繁忙、不愿惹麻煩等原因,漏報、遲報現象較為嚴重,同時,不良反應報告質量也不理想,無法及時提煉出嚴重的、緊急的有效風險信息[20]。因此,建議我國藥品監督管理部門參考FDA的Medwatch自愿報告系統,針對消費者單獨設計不良反應報告表格,允許消費者在線填報不良反應,這一舉措符合我國國情,并且從維護人民用藥安全、促進合理用藥的角度來看是迫切需要的。目前我國現代信息技術發展迅速,并且有美國Medwatch系統運行的經驗,通過電話、網站、官方手機應用軟件等方式來擴大報告來源具有一定的可行性。
3.2 建立統一的數據收集、存儲系統和科學的數據處理方法
信息的質量決定了藥品風險管理能否及時發現藥品風險信號,進而采取措施控制或預防用藥風險。對此,美國的不良反應監測已由收集病例階段發展到對病例的評價利用階段,信息系統的應用為不良反應監測提供了有力的技術支持。
我國于2012年建立藥品不良反應監測系統,并且運用報告比數比法(ROR)、英國藥品和保健產品管理局(MHRA)使用的綜合標準法、比例報告比值比法(PRR)、貝葉斯可信傳播神經網絡法(BCPNN)四種方法檢測數據庫。目的是為了能及時監測到藥品上市后安全信息,通過數據分析,對可能出現的不良反應及時做出預防措施。我國藥品安全監測目前仍采用被動監測模式,這種模式已表現出一些不足之處[21]。因此,我國應完善基于公共數據庫模型的藥品安全監測系統,對風險較高的藥品建立主動監測模式,降低藥品安全問題發現的滯后性,形成主動與被動監測相補充的監測系統,這也符合我國國情。相關部門應充分利用醫療機構的數據資源,將已有醫院信息系統(Hospital information system,HIS)中的電子處方記錄等數據進行連接整合,采用統一術語集進行數據標準化,將包含大量藥品安全信息的臨床醫療數據和藥品使用記錄應用于藥品安全監測。此外,還應擴大不同利益相關方的權限,使監管部門、企業、醫療機構等多方能夠獲取并分享數據庫中信息,監管部門要做好數據集中處理工作,并且探索更高效的數據挖掘手段與評估方式,以便更及時地獲取不良反應信息。2011年,我國與世界衛生組織(WHO)的數據庫進行交流與對接,足以證明進一步完善數據庫建設具有可行性。
3.3 不斷強化制藥企業監測、報告的主體責任意識
企業應當是藥品安全的第一責任人與不良反應監測的責任主體。美國制藥企業在FDA的嚴格監管、醫藥產業發展和自身責任意識提高等因素影響下,已形成相對完備的不良反應報告和監測管理制度。美國實施交叉上報系統,藥品不良反應的上報者在向不良反應監測部門上報的同時向藥品生產企業通報;或藥品不良反應監測部門接到報告后在報告者允許的范圍內向生產企業報送;又或藥品生產企業收到報告者的不良事件報告后再向藥品不良反應監測部門報告。在這種體系下,藥品生產企業獲知不良反應的概率大幅增加,為企業的主動上報奠定了基礎。另一方面,FDA能夠以交叉上報情況為依據,對已經接收到不良反應報告但未主動上報給FDA的生產企業進行重點監察,間接督促企業主動開展不良反應監測和報告工作。企業主動性和責任意識的增強,能夠在直接保障患者用藥安全的同時,進一步提升了企業的自我檢查和產品風險控制能力。
我國目前已經有對藥品生產企業進行風險管理的法律規定,也發布藥品上市后監測的指南文件,如2015年9月10日,國家食品藥品監督管理總局發布的《藥品不良反應報告和監測檢查指南(試行)》。與醫療機構所提供的報告數相比,生產企業不良反應報告數非常低,作用十分有限[22]。分析內在原因,企業可能本身對報告不夠重視,也有可能欠缺獲取不良反應數據的渠道,因此無法形成報告,此外,部分企業因為理念上對不良反應有所誤解,擔心上報不良反應會影響企業的業績與名譽,因此缺乏上報動力。加大對藥品上市后的安全關注力度,是推進我國合理用藥、保障人民用藥安全的重要環節。因此,我國應進一步強調制藥企業的主體意識,提高企業、政府、患者與醫療機構的溝通效力與合作意識。應通過加強約束力,完善監督隊伍建設以及加大監督力度,并且加強宣傳培訓的手段,使企業主動承擔藥品安全與不良反應監測的主體責任[23]。2018年,我國“藥品上市許可持有人藥品不良反應直接報告系統”正式出臺,藥品生產企業將通過該系統上報不良反應,可見我國已經將強化企業主體責任向具體實踐方面探索,進一步增強對藥品不良反應的監測、預防與控制。
4 結語
我國在著力加快藥品上市審評速度的同時,應當保障再評價體系完善,達到鼓勵創新與保障安全的雙重目標。在FDA再評價體系的研究中,我國可以借鑒美國以企業作為責任主體,對上市后藥品不良反應進行主動監測,拓寬不良反應投訴與報告渠道的理念,推動再評價體系的持續完善,不斷降低患者用藥風險。
猜你喜歡
車主之友(2022年6期)2023-01-30 08:01:04
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
車主之友(2022年4期)2022-11-25 07:27:30
車主之友(2022年4期)2022-08-27 00:57:48
南方人物周刊(2017年32期)2017-10-28 22:48:36
南風窗(2016年26期)2016-12-24 21:48:09
中國衛生(2016年5期)2016-11-12 13:25:28
中國衛生(2015年5期)2015-11-08 12:09:48
南風窗(2015年22期)2015-09-10 07:22:44
南風窗(2015年7期)2015-04-03 01:21:48
