UPLC法測定射干藥材中10個異黃酮類成分的含量
2019-09-10 01:22:27姜鴻王光函辛旭陽鄒桂欣李國信
中國藥房 2019年23期
姜鴻 王光函 辛旭陽 鄒桂欣 李國信




中圖分類號 R284.2 文獻標志碼 A 文章編號 1001-0408(2019)23-3216-05
DOI 10.6039/j.issn.1001-0408.2019.23.09
摘 要 目的:建立同時測定射干藥材中10個異黃酮類成分含量的測定方法,并用于評價不同產地射干藥材中各成分的含量差異。方法:采用超高效液相色譜(UPLC)法。色譜柱為Waters ACQUITY UPLC BEH C18,流動相水相組成為含0.5%甲基-β-環糊精、0.1%磷酸的水溶液,有機相為乙腈,梯度洗脫,流速為0.2 mL/min,柱溫為35 ℃,檢測波長為265 nm,進樣量為2 μL,分析時間為20 min。對來自于8個省份的26個樣品中的10個異黃酮類成分射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素進行含量測定。結果:射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素檢測質量濃度線性范圍分別為8.569 5~342.78、0.643~25.72、1.119 8~44.79、2.187 8~87.51、0.770 3~30.81、0.421 3~16.85、0.288 5~11.54、1.795 3~71.81、0.560 8~22.43、0.086~3.44 μg/mL(r均≥0.999 6),定量限分別為0.015、0.102、0.096、0.013、0.036、0.088、0.102、0.019、0.067、0.092 μg/mL;精密度、穩定性(24 h)、重復性試驗的RSD均<2.00%(n=6);加樣回收率為95.30%~103.30%(RSD均≤2.33%,n=6)。在26個射干樣品中,以射干苷含量最高(3.66%~57.79%),白射干素含量最低(0.09%~0.59%),次野鳶尾黃素含量為0.29~2.80 mg/g,且不同產地中各異黃酮類成分含量差異較大。結論:建立的含量測定方法靈敏、分析時間較短、重復性較好,可以用于同時測定射干藥材中10個異黃酮類成分的含量及評價各成分含量差異。
關鍵詞 射干;不同產地;異黃酮類成分;含量測定;超高效液相色譜法
Content Determination of 10 Isoflavones in Belamcanda chinensis by UPLC
JIANG Hong,WANG Guanghan,XIN Xuyang,ZOU Guixin,LI Guoxin(Medical Research Institute of Liaoning Province, Shenyang 110034, China)
ABSTRACT ? OBJECTIVE: To establish a method for simultaneous determination of 10 isoflavones in Belamcanda chinensis, and to evaluate the differences of active ingredient content of B. chinensis from different areas. METHODS: UPLC method was adopted. The determination was performed on Waters ACQUITY UPLC BEH C18 column with mobile phase consisted of 0.5 % methyl-β-cyclodextrin and 0.1% phosphate as water phase, acetonitrile as organic phase (gradient elution) at the flow rate of 0.2 mL/min. The column temperature was set at 35 ℃, and the detection wavelength was set at 265 nm. The sample size was 2 μL, and analysis time was 20 min. The contents of 10 isoflavones in 26 samples from 8 provinces, including tectoridin, iristectorin A, iristectorin B, iridin, tectorigenin, iristectorigenin B, iristectorigenin A, irigenin, irisflorentin, dichotomitin, were determined. RESULTS: The linear ranges of tectoridin, iristectorin A, iristectorin B, iridin, tectorigenin, iristectorigenin B, iristectorigenin A, irigenin, irisflorentin, dichotomitin were 8.569 5-342.78, 0.643-25.72, 1.119 8-44.79, 2.187 8-87.51, 0.770 3-30.81, 0.421 3- 16.85, 0.288 5-11.54, 1.795 3-71.81, 0.560 8-22.43, 0.086-3.44 μg/mL(all r≥0.999 6). The limits of quantitation were 0.015, 0.102, 0.096, 0.013, 0.036, 0.088, 0.102, 0.019, 0.067, 0.092 μg/mL. RSDs of precision, stability(24 h)and reproducibility tests were lower than 2.00% (n=6). The recoveries ranged 95.30%-103.30% (all RSD≤2.33%, n=6). Among 26 samples of B. chinensis, the content of tectoridin was the highest (3.66%-57.79%), and the content of dichotomitin was the lowest (0.09%- 0.59%), the contents of irisflorentin were 0.29-2.80 mg/g. The contents of isoflavones in B. chinensis from different areas were different greatly. ?CONCLUSIONS: The established method is sensitive, with short analysis time and good repeatability, and can be used to determine the content of 10 isoflavones and evaluate the content difference of each component.
KEYWORDS ? Belamcanda chinensis; Different producing areas; Isoflavone; Content determination; UPLC
射干為鳶尾科植物射干[Belamcanda chinensis(L.)DC.]的根莖,具有清熱解毒、消痰、利咽的功效,主要用于咽喉腫痛、痰涎壅盛、咳嗽氣喘[1],為臨床常用藥材。大量研究表明,射干含有大量黃酮和異黃酮類化合物,其中的異黃酮類化合物具有抗炎、抗病毒、抗氧化、抑菌、止咳、抗癌、類雌激素作用等藥理活性[2-7]。射干藥材為多年生草本植物,生長和種植范圍很廣,各產地或同一產地不同生長年限、不同采收季節射干藥材中異黃酮類成分含量差異較大[8-10]。目前,2015年版《中國藥典》(一部)[1]中射干藥材只是以次野鳶尾黃素含量標定藥材質量,而中藥的藥理作用是多種化學成分綜合作用的結果,單一或少數幾個指標性成分無法代表中藥材的總體質量。因此,建立一個科學的射干藥材質量評價方法具有重要的科學意義。
筆者查閱有關射干和鳶尾屬藥材的含量測定文獻,已有研究者建立了可以同時測定射干或鳶尾屬藥材中6或7個異黃酮類成分含量的方法[11-12],但筆者采用這些方法欲同時測定射干中的10個異黃酮類成分(射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素)時,發現部分峰之間不易分開,分離效果不好。由于射干中的上述異黃酮類成分中有很多化學結構比較相似或接近,如鳶尾甲苷A、B及鳶尾甲黃素A、B分別互為同分異構體,而野鳶尾黃素在結構上也只比鳶尾甲黃素A多一個甲氧基。因此,采用常規的高效液相色譜(HPLC)法在C18柱上很難實現上述所有成分的有效分離。筆者曾采用梯度洗脫方法,耗時將近4 h才能勉強將各成分有效分離,但大部分色譜峰峰形較差。之后,筆者通過在流動相中添加甲基-β-環糊精,采用超高效液相色譜(UPLC)法,發現可在20 min內(若采用HPLC法,分析時間為60 min)將射干中上述10個異黃酮類成分完全分離。最終,筆者成功建立了同時測定射干中10個異黃酮類成分含量的方法,并采用此法對不同產地、不同生長年限、不同采收季節的射干藥材的異黃酮類成分含量進行了測定及比較,以期為合理選擇產地藥材提供參考。
1 材料
1.1 儀器
UPLC儀、全自動進樣器、二極管陣列檢測器(美國Waters公司);電子天平(瑞士梅特勒-托利多儀器有限公司);KQ3200超聲波清洗器(昆山市超聲儀器有限公司,本文試驗所用超聲功率均為120 W,工作頻率均為40 kHz)。
1.2 藥品與試劑
射干苷對照品(批號:111632-200501,純度:>98%)、次野鳶尾黃素對照品(批號:111557-200602,純度:>98%)均購于中國食品藥品檢定研究院;野鳶尾苷對照品(批號:13030803,純度:>93%)、野鳶尾黃素對照品(批號:130514023,純度:>98%)均購于成都普瑞法科技公司;鳶尾黃素對照品(批號:12081808,純度:>98%)購于成都生物科技有限公司;鳶尾甲苷A對照品(批號:100287)、鳶尾甲苷B對照品(批號:100763)、鳶尾甲黃素A對照品(批號:20140724)、鳶尾甲黃素B對照品(批號:20140616)、白射干素對照品(批號:20140621),純度:均>98.5%,均購于江蘇永健醫藥科技有限公司;磷酸、乙腈均為色譜純;水為屈臣氏蒸餾制法飲用水(批號:20190223);甲基-β-環糊精(美國Aladdin公司,購于國藥集團化學試劑沈陽有限公司,平均分子量:1 310,批號:G1529014);各不同產地、不同季節采集或購買的射干藥材,經遼寧省中醫藥研究院藥學研究室王光函研究員鑒定,均為鳶尾科植物射干[Belamcanda chinensis(L.)DC],樣品產地及采集或購買日期信息見表1(部分產地具體信息不明)。
2 方法與結果
2.1 色譜條件
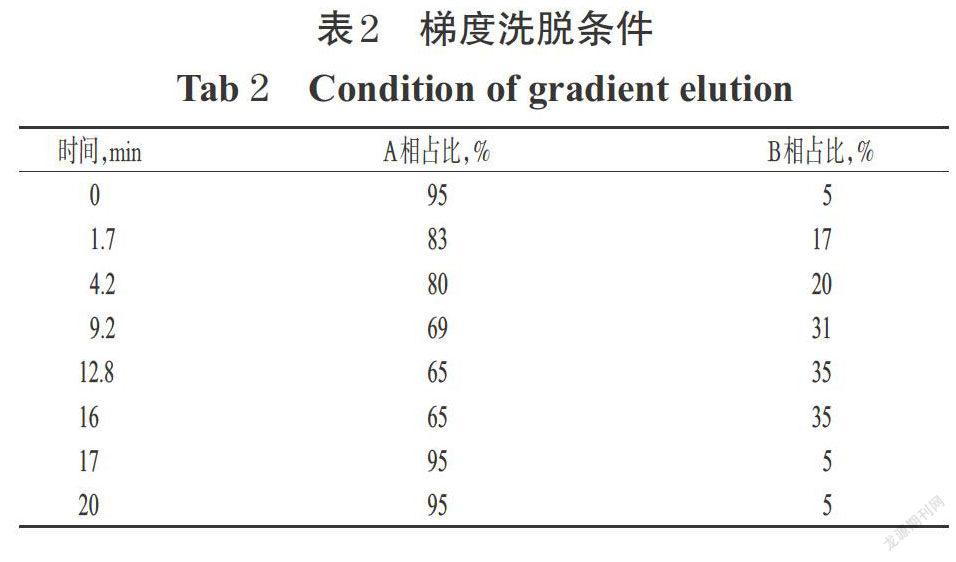
色譜柱為Waters ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm);流動相A相(水相)為水溶液(含0.5%甲基-β-環糊精、0.1%磷酸),B相(有機相)為乙腈,梯度洗脫,流速為0.2 mL/min;柱溫為35 ℃;檢測波長為265 nm;進樣量為2 μL,分析時間為20 min。梯度洗脫條件見表2。
2.2 溶液制備
2.2.1 混合對照品溶液制備 精密稱取射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素對照品適量,加甲醇制成質量濃度依次為342.78、25.72、44.79、87.51、30.81、16.85、11.54、71.81、22.43、3.44 μg/mL的混合對照品溶液。
2.2.2 供試品溶液制備 取射干藥材適量,粉碎成細粉;精密稱取0.1 g,精密加入70%乙醇25 mL,超聲提取30 min,放至室溫,用70%乙醇補足減失質量,用0.22 ? ?μm微孔濾膜濾過,取續濾液適量即得。
2.3 系統適用性試驗
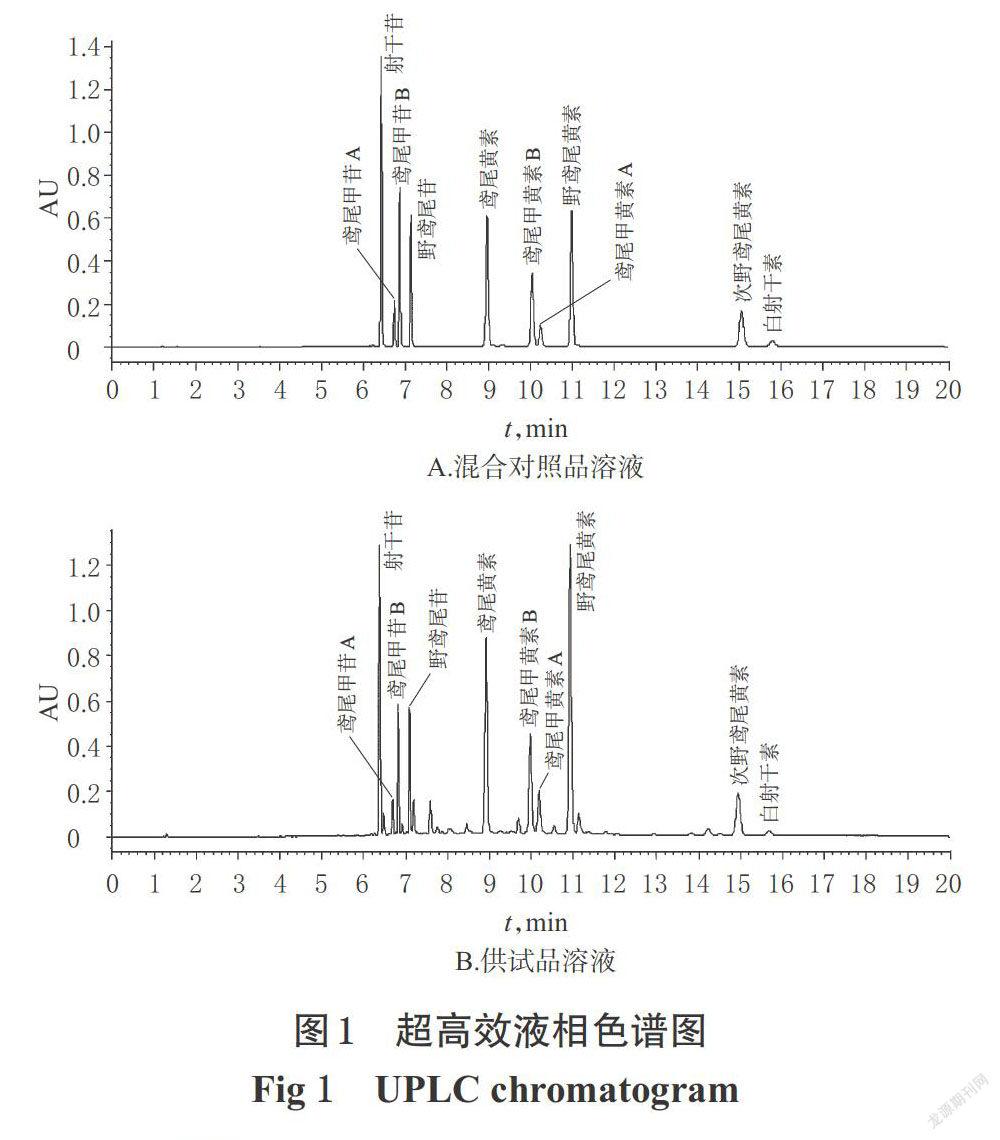
取“2.2.1”“2.2.2”項下混合對照品溶液、供試品溶液,分別進樣2 μL,測定。結果,在該色譜條件下,各成分色譜峰均能達到基線分離,且相鄰兩峰之間分離度均大于1.5,10個成分色譜峰的理論板數均不低于3 000。色譜圖見圖1。
2.4 方法學考察
2.4.1 線性關系考察 分別精密吸取“2.2.1”項下混合對照品溶液0.125、0.25、0.5、2、2.5 mL,分別置于5 mL量瓶中,加甲醇稀釋至刻度,搖勻,制備成包含原液在內的共6個質量濃度的系列混合對照品溶液。分別精密吸取各質量濃度的對照品混合溶液進樣測定。以各色譜峰面積(y)為縱坐標、相應的對照品溶液質量濃度(x)為橫坐標進行線性回歸,得回歸方程及線性范圍,結果見表3。
2.4.2 定量限考察 精密量取“2.2.1”項下混合對照品溶液適量,倍比稀釋后測定,以信噪比為10 ∶ 1計算定量限。結果,射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素的定量限依次為 0.015、0.102、0.096、0.013、0.036、0.088、0.102、0.019、0.067、0.092 μg/mL。
2.4.3 精密度試驗 取“2.2.2”項下供試品溶液(樣品編號10)適量,連續進樣測定6次,記錄峰面積。結果,射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素峰面積的RSD依次為0.38%、0.52%、0.33%、0.35%、0.72%、0.67%、1.15%、1.02%、0.51%、1.53%(n=6),表明本測定方法精密度良好。
2.4.4 穩定性試驗 取“2.2.2”項下供試品溶液(樣品編號10)適量,分別于室溫下放置0、2、4、8、12、24 h時,按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素峰面積的RSD依次為0.56%、0.71%、0.39%、0.28%、0.66%、0.93%、1.21%、1.39%、0.59%、1.72%(n=6),表明供試品溶液于室溫下放置24 h內基本穩定。
2.4.5 重復性試驗 精密稱取樣品(樣品編號10)粉末適量,共6份,按“2.2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算各待測成分含量。結果,射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素的含量依次為14.09、1.19、2.85、3.65、3.45、1.94、1.36、6.46、1.62、0.36 mg/g;含量的RSD依次為0.66%、1.23%、1.05%、0.87%、0.69%、1.77%、1.58%、0.62%、1.11%、1.85%(n=6),表明本含量測定方法重復性良好。
2.4.6 回收率試驗 取已知含量的樣品(樣品編號10)粉末0.05 g,共6份,精密稱定,分別加入混合對照品溶液(射干苷、鳶尾甲苷A、鳶尾甲苷B、野鳶尾苷、鳶尾黃素、鳶尾甲黃素B、鳶尾甲黃素A、野鳶尾黃素、次野鳶尾黃素、白射干素對照品適量,加甲醇制成質量濃度依次為702.8、58.26、143.35、181.62、175.33、97.37、68.62、325.18、80.92、18.17 μg/mL)的溶液1 mL,按“2.2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算回收率,結果見表4。
2.5 樣品含量測定
取來自于8個省份的26個樣品,按“2.2.2”項下制備成供試品溶液,按“2.1”項下色譜條件進樣測定,結果見表5。
3 討論
3.1 環糊精作為流動相添加劑的分離機制
環糊精可用于手性化合物的異構體、對映體的分離,一般的化合物分子能夠嵌入環糊精內腔并發生包絡作用,但由于各化合物分子結構不同,于是在包結絡合差異上較大,由此為不同結構化合物的分離產生了條件。也有些化合物,其碳原子上的羥基能夠與環糊精腔體上的羥基結合成氫鍵產生相互作用,由于羥基位置不同,因而使得化合物之間的色譜行為差異擴大,并提高分離度[13-14]。
β-環糊精及其衍生物以其獨特的分子結構、尺寸和性能,常被作為流動相的手性添加劑,這樣即使采用常規色譜柱也可實現手性化合物與同分異構體的分離,具有使用方便、不干擾檢測信號、分離效果好、分析成本低等優點。目前,β-環糊精及其衍生物已有多種商品化產品,筆者在前期試驗中曾考察了β-環糊精、甲基-β-環糊精、羥丙基-β-環糊精、三甲基-β-環糊精環糊精、羧甲基-β-環糊精對射干中10個異黃酮化合物分離的影響,最后綜合考慮各色譜峰的分離效果,發現用甲基-β-環糊精作為流動相添加劑效果。
3.2 甲基-β-環糊精濃度對分離的影響
在前期研究中,筆者曾考察了流動相中分別添加0.1%、0.25%、0.5%、1%等不同濃度甲基-β-環糊精的分離效果,發現隨著甲基-β-環糊精濃度的提高,容量因子迅速降低,分離度增大。但甲基-β-環糊精濃度太高時,由于溶解度的限制,會影響其在流動相中的溶解,使流動相變渾濁。綜合考慮,以水相中含0.5%的甲基-β-環糊精較為合適,在此條件下各色譜峰均能達到良好的分離。
3.3 流動相有機改性劑對分離的影響
在前期試驗中,選用有機改性劑時分別選擇了甲醇和乙腈,觀察供試品溶液中各成分的分離情況。結果發現,當有機相使用乙腈時,各成分均能達到良好的分離,而且基線沒有漂移現象,各成分色譜峰峰形良好。
3.4 流動相中磷酸的加入對分離的影響
筆者在本研究中發現,當流動相水相中加入0.1%的磷酸時,可以使各成分的色譜峰峰形有很大改善,同時也可以改善分離度,且磷酸的加入沒有造成基線漂移現象。
3.5 柱溫對分離的影響
筆者在本研究中發現,隨著色譜柱柱溫的升高,各色譜峰峰形變好,分析時間變短,但有些成分的分離度也隨著減小。這可能是因為溫度對各成分的影響不一致,或者柱溫過高時,有些化合物與環糊精形成的絡合物的穩定性變差了,從而降低了手性分離作用。因此,綜合考慮分離度、色譜峰形和保留時間等因素,最終選擇35 ℃作為色譜柱的溫度。
3.6 多成分分析評價中藥材質量
中藥材成分復雜,具有多成分、多靶點、多途徑、整體協同作用的特點,一般的單一化學成分不能完全代表藥材的真實質量。在本研究中,筆者采用了同時測定不同成分的分析評價方法,此法可為中藥質量評價提供有益的思路和方法,能夠較全面地分析藥材質量,為中藥藥材質量綜合評價提供了參考,具有較好的實際應用價值。
3.7 不同產地及采收期對藥材有效成分含量的影響
一般來說,藥材所含的有效成分在生長發育的不同時期含量各不相同,由此可導致藥物臨床療效及副作用方面的差異。唐朝孫思邈在《千金要方》中云:“早則藥勢未成,晚則盛時已歇”[15];明朝陳嘉謨在《本草蒙筌》中總結出中藥采制的原則,并專列出“出產擇地土”“采收按時月”“凡諸草、木、蟲產之有地,根、葉、花、實采之有時。失其地,則性味少異,失其時,則氣味不全”[16]。上述論著均強調了藥材產地和適時采收的重要性,且藥材質量受其生長地的氣候、土壤等條件的影響也非常大。
從收集的各產地射干藥材的含量測定結果可以看出,異黃酮類成分含量差異較大,但次野鳶尾黃素含量為0.29%~2.80%(mg/g),符合2015年版《中國藥典》(一部)[1]中>0.10%的標準。苷類成分由南向北有減少的趨勢,尤其是射干苷更為明顯;而苷元類成分,以鳶尾黃素、野鳶尾黃素為代表,由南向北略有增加趨勢。從10個異黃酮類成分含量均衡性看,湖北黃岡市團風縣射干良好農業規范基地、河南南陽桐柏的射干藥材較好,各成分含量相對均衡,更符合多成分、多靶點、多途徑整體協同作用的中醫藥特點。由于這些成分具有抗病毒、抗炎、抑菌、止咳、抑制腫瘤細胞、類雌激素作用等藥理活性[4-7],因此,有必要對射干進一步進行系統研究,確認其中發揮不同藥理作用的相應活性成分,以便在臨床應用時,根據藥材中各活性成分含量情況,選擇不同產地射干藥材,用于不同的臨床癥狀,使其更能充分發揮臨床療效。
參考文獻
[ 1 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2015年版.北京:中國醫藥科技出版社,2015:285.
[ 2 ] 羅森,袁崇均,陳帥,等.川射干葉的化學成分研究[J].中國藥房,2016,27(30):4267-4269.
[ 3 ] 張杰,曾鋮,常義生,等.射干化學成分研究[J].安徽農業科學,2015,43(24):57-59.
[ 4 ] 王振飛,劉麗,陳永霞,等.射干提取物抑制肺癌細胞惡性行為的研究[J].國醫論壇,2018,33(2):57-59.
[ 5 ] 徐倩.射干不同有效成分體外抗病毒藥效學作用分析[J].亞太傳統醫藥,2015,11(18):9-10.
[ 6 ] 張琳,張妮.川射干的化學及藥理研究進展[J].陜西中醫學院學報,2014,37(5):91-93.
[ 7 ] 溫雯,馬躍海,朱竟赫,等.射干傳統功效考證及其實驗藥理學驗證[J].世界科學技術:中醫藥現代化,2017,19(5):846-850.
[ 8 ] 普冰清,朱艷,許方云,等.射干不同采收期黃酮類成分的動態變化規律[J].藥學與臨床研究,2014,22(3):212-215.
[ 9 ] 李霞,崔月曦.射干質量的影響因素[J].中國醫藥指南,2014,12(18):289-290.
[10] 陳帥,袁崇均,羅森,等.不同產地川射干中異黃酮成分含量比較研究[J].藥物分析雜志,2018,38(7):1280-1284.
[11] 卞婭,劉孟生,張麗媛,等.射干、鳶尾不同部位6種活性成分定量分析及抗炎作用初探[J].中國中藥雜志,2018,43(1):119-122.
[12] 陳帥,袁崇均,羅森,等.超高效液相色譜法同時測定川射干中7個異黃酮成分的含量[J].天然產物研究與開發,2018,30(2):256-260,217.
[13] 徐雄,寧衛紅,瞿志榮,等. HPLC手性流動相添加劑及其用于藥物對映體拆分中的影響因素[J].生物化工,2018,4(5):150-155.
[14] 鄭振,陳秀娟,趙亮,等.衍生化β-環糊精手性固定相高效液相色譜法拆分米那普侖對映體及其分離機制[J].色譜,2017,35(3):286-290.
[15] 孫思邈.千金方[M].北京:中國中醫藥出版社,1998:18.
[16] 陳嘉謨.本草蒙筌[M].北京:人民衛生出版社,1988:1-2.
(收稿日期:2019-07-26 修回日期:2019-09-15)
(編輯:劉 萍)
