基于HPLC指紋圖譜及UPLC-Q-TOF-MS法的3種不同來源大黃差異成分研究
2019-09-10 01:22:27李丹丹張慧李思雨郭泰麟
中國藥房 2019年23期
李丹丹 張慧 李思雨 郭泰麟




中圖分類號 R284 文獻標志碼 A 文章編號 1001-0408(2019)23-3240-06
DOI 10.6039/j.issn.1001-0408.2019.23.13
摘 要 目的:建立不同來源大黃藥材的高效液相色譜(HPLC)指紋圖譜,并對其中差異成分進行定性鑒別。方法:采用“中藥色譜指紋圖譜相似度評價系統”(2012版)軟件建立30批大黃藥材(掌葉大黃20批、唐古特大黃和藥用大黃各5批)的HPLC指紋圖譜并進行相似度評價,采用偏最小二乘判別法(PLS-DA)結合超高效液相色譜(UPLC)-四極桿-飛行時間串聯質譜(Q-TOF-MS)法定性鑒別3種來源大黃的差異性成分。結果:30批大黃藥材樣品指紋圖譜的相似度在0.609~0.960之間。經PLS-DA分析,大黃藥材樣品按來源明顯聚集為3組,3組樣品間差異性成分共有18個,經UPLC-Q-TOF-MS鑒別為白藜蘆醇-4′-O-β-D-(6″-O-沒食子酰)-葡萄糖苷、蓮花掌苷、大黃酸-8-O-葡萄糖苷、表兒茶素沒食子酸酯、4-(4′-羥苯基)-2-丁酮-4′-O-β-D-(2″-O-肉桂酰基-6″-O-沒食子酰基)-葡萄糖苷等。結論:建立的方法可有效區分3種不同來源大黃藥材,并對其差異成分進行鑒別,為多來源藥材的鑒別和質量評價提供了參考。
關鍵詞 大黃;指紋圖譜;高效液相色譜;超高效液相色譜-四極桿-飛行時間-串聯質譜法;偏最小二乘判別法;差異成分
Study on Different Components of Rhei Radix Et Rhizoma from 3 Different Origins Based on HPLC Fingerprint and UPLC-Q-TOF-MS
LI Dandan,ZHANG Hui,LI Siyu,GUO Tailin(College of Pharmacy,Liaoning University of TCM,Liaoning Dalian 116600,China)
ABSTRACT ? OBJECTIVE: To establish HPLC fingerprints of Rhei Radix Et Rhizoma from different origins, and identify the differential components qualitatively. METHODS: HPLC fingerprints of 30 batches of Rhei Radix Et Rhizoma (20 batches of Rheum palmatum, 5 batches of Rheum tanguticum and 5 batches of Rheum officinale) were established and similarity evaluation was performed by using Similarity Evaluation System for Chromatographic Fingerprint of TCM (2012 edition). Qualitative identification of differential components of Rhei Radix Et Rhizoma from 3 different origins were performed by using PLS-DA combined with UPLC-Q-TOF-MS. RESULTS:The fingerprint similarities of 30 batches of samples were between 0.609 and 0.960. According to PLS-DA analysis, Rhei Radix Et Rhizoma were significantly aggregated into 3 groups according to the origin. There were 18 different components among 3 groups, which were identified by UPLC-Q-TOF-MS as resveratrol-4′-O-β-D-(6″-O-gallacyl)-glucoside, lindleyin, rhein-8-O-glucoside, epicatechin gallate, 4-(4′-hydroxyphenyl)-2-butanone-4′-O-β-D-(2″-O-cinnamyl-6″-O-gallacyl)-glucoside. CONCLUSIONS: Established method can effectively identify Rhei Radix Et Rhizoma from 3 different origins, and the differential components can be distinguished, which provides a reference for the identification and quality evaluation of multi-source medicinal materials.
KEYWORDS ? Rhei Radix Et Rhizoma; Fingerprint; HPLC; UPLC-Q-TOF-MS; PLS-DA; Differential component
大黃為蓼科植物掌葉大黃(Rheum palmatum L.)、唐古特大黃(Rheum tanguticum Maxim.ex Balf.)或藥用大黃(Rheum officinale Baill.)的干燥根和根莖,具有瀉下攻積、清熱瀉火、涼血解毒、逐瘀通經等功效[1],是臨床常用中藥,且用藥歷史悠久。現今,在市場上流通的3種正品大黃藥材和飲片質量參差不齊,行業內公認唐古特大黃和掌葉大黃為優良品種[2]。但3者在外觀性狀上相似,通過性狀難以實現有效區分,因此市場上常見大黃品種來源混淆的現象[3]。為此,亟需建立區分3種來源大黃的鑒別方法,同時尋找出3種大黃的差異性成分,以控制大黃藥材的質量。目前,雖有大黃藥材指紋圖譜以及3種大黃藥材預測模型構建等方面的研究[4-5],但3種來源大黃的差異性成分仍不清楚。基于此,本研究結合化學計量學的方法[偏最小二乘判別法(PLS-DA法)],對3種不同來源大黃的全成分指紋圖譜進行對比分析,篩選出差異成分峰,并利用超高效液相色譜(UPLC)-四極桿-飛行時間串聯質譜(Q-TOF-MS)法對差異成分峰進行定性識別,從而明確并識別3種來源大黃的差異成分,為大黃藥材的質量控制和綜合利用提供科學依據。
1 材料
1.1 儀器
CP225D電子天平(德國賽多利斯科學儀器有限公司);KQ-250D數控超聲波清洗器(昆山超聲儀器有限公司);FW-80小型中藥高速粉碎機(天津泰斯特儀器有限公司);TDZ4-WS低速臺式離心機(湖南湘儀實驗室儀器開發有限公司);6550 Q-TOF-MS儀、1260高效液相色譜儀、1290 UPLC儀、G4212B 1260二極管陣列檢測器(DAD)(美國安捷倫公司)。
1.2 藥品與試劑
30批藥材收集于四川、甘肅、青海等省,經遼寧中醫藥大學藥學院張慧教授鑒定為蓼科植物掌葉大黃(R. palmatum L.)、唐古特大黃(R. tangguticum Maxim.ex Balf.)和藥用大黃(R. officinale Balf.)的干燥根和根莖;蘆薈大黃素-8-O-葡萄糖苷對照品(批號:CHB190115,純度:≥98%)、大黃酚-8-O-葡萄糖苷對照品(批號:CHB180726,純度:≥98%)、大黃素-8-O-葡萄糖苷對照品(批號:CHB180727,純度:≥98%)、大黃酸-8-O-葡萄糖苷對照品(批號:CHB180726,純度:≥98%)、番瀉苷A對照品(批號:CHB180729,純度:≥98%)均由成都克洛瑪生物科技有限公司提供;沒食子酸對照品(上海源葉生物科技有限公司,批號:Y19M8C36143,純度:≥98%);大黃素對照品(批號:110756-200110,純度:≥98.6%)、大黃素甲醚對照品(批號:110758-201013,純度:≥99.8%)、大黃酸對照品(批號:110757-200206,純度:≥99.8%)、蘆薈大黃素對照品(批號:110795- 200806,純度:≥98.6%)均由中國食品藥品檢定研究院提供;甲醇、甲酸為色譜純,水為純凈水。大黃樣品來源信息見表1。
2 方法與結果
2.1 色譜條件
色譜柱:Phenomenex C18(250 mm ×4.6 mm,5 μm);流動相:甲醇(A)-0.4%甲酸水(B),梯度洗脫(0~8 min,5%→30%A;8~15 min,30%→33%A;15~25 min,33%→35%A;25~30 min,35%→37%A;30~35 min,37%→40%A;35~47 min,40%→45%A;47~64 min,45%→50%A;64~80 min,50%→55%A;80~83 min,55%→60%A;83~93 min,60%→100%A);流速:1.0 mL/min;檢測波長:254 nm;柱溫:30 ℃;進樣量:10 μL。
2.2 供試品溶液的制備
將大黃藥材粉碎過80目篩,取粉末0.5 g,精密稱定,置于具塞錐形瓶中,精密加入甲醇25 mL,稱定質量,室溫超聲(頻率:40 kHz,功率:200 W)30 min,取出放冷,再稱定質量,用甲醇補足減失的質量,離心 (4 000 ? r/min) 5 min,取上清液過0.22 μm微孔濾膜,即得。
2.3 對照品貯備液的制備
精密稱取沒食子酸、蘆薈大黃素-8-O-葡萄糖苷、大黃酸-8-O-葡萄糖苷、番瀉苷A、大黃酚-8-O-葡萄糖苷、大黃素-8-O-葡萄糖苷、蘆薈大黃素、大黃酸、大黃素、大黃素甲醚對照品適量,分別置于不同的10 mL量瓶中,用甲醇稀釋至刻度,制備成質量濃度分別為0.146、0.600、0.506、0.556、0.600、0.407、0.130、0.353、0.103、1.066 ? ?mg/mL的對照品貯備液,4 ℃保存,備用。
2.3 方法學考察
2.3.1 精密度試驗 取S9號樣品,按“2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件連續進樣6次,記錄色譜圖。以峰4(蘆薈大黃素-8-O-葡萄糖苷峰)為參照峰,計算得各共有峰相對保留時間的RSD均小于1.5%(n=6)、相對峰面積的RSD均小于3.0%(n=6),表明儀器的精密度良好。
2.3.2 穩定性試驗 取S9號樣品,按“2.2”項下方法制備供試品溶液,在室溫放置0、3、6、9、12、24 h后,分別按“2.1”項下色譜條件進樣,記錄色譜圖。以峰4(蘆薈大黃素-8-O-葡萄糖苷)為參照峰,計算得各共有峰相對保留時間的RSD均小于1.5%(n=6)、相對峰面積的RSD均小于5.0%(n=6),表明樣品在室溫條件下24 h內穩定性良好。
2.3.3 重復性試驗 取S9號樣品,按“2.2”項下方法平行制備6份供試品溶液,分別按“2.1”項下色譜條件進樣,每份樣品進樣2次,記錄色譜圖。以峰4(蘆薈大黃素-8-O-葡萄糖苷)為參照峰,計算得各共有峰相對保留時間的RSD均小于1.5%(n=6)、相對峰面積的RSD均小于5.0%(n=6),表明該方法重復性良好。
2.4 指紋圖譜的建立及分析
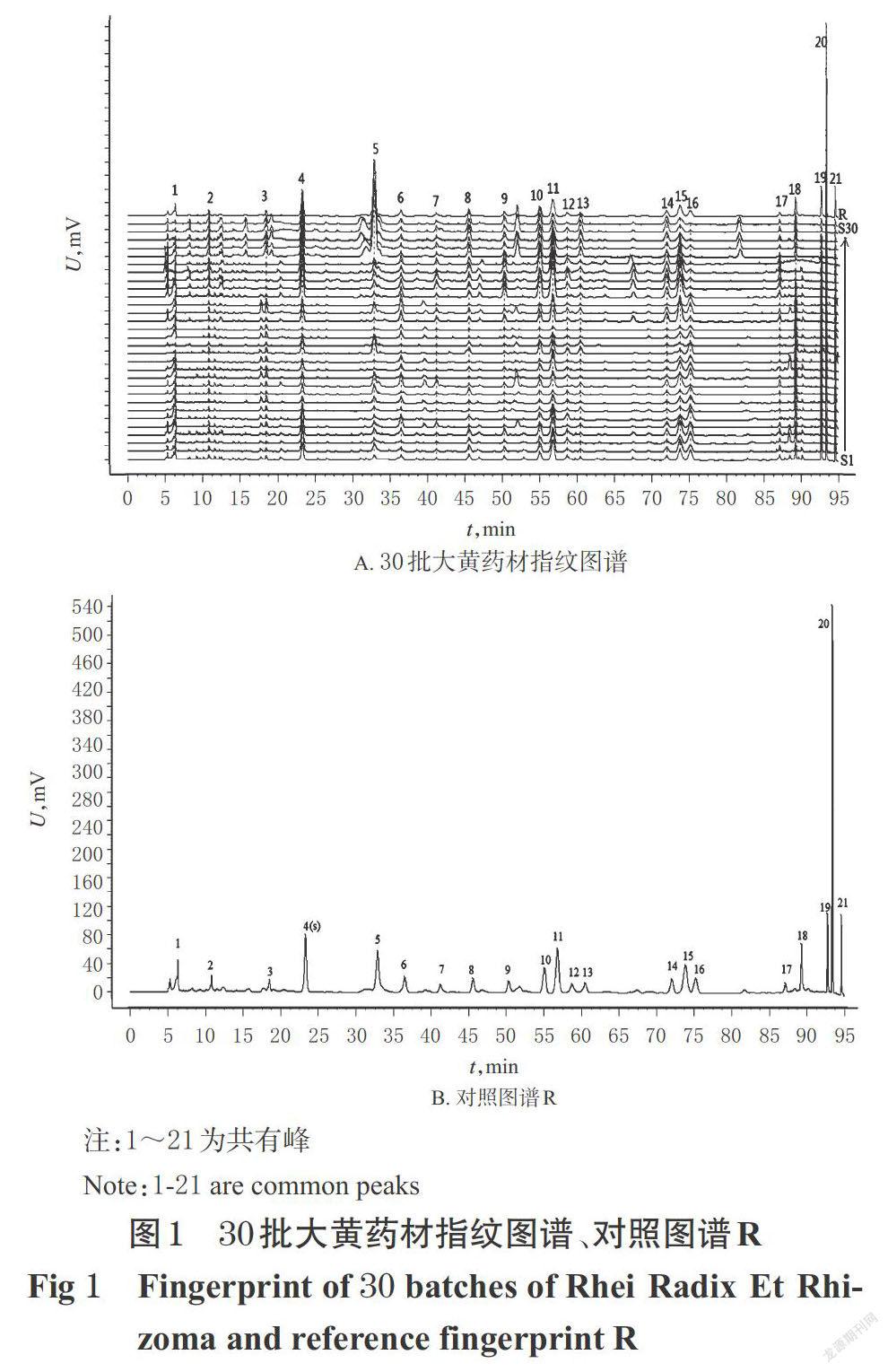
2.4.1 30批大黃指紋圖譜的建立及相似度評價 按“2.2”項下方法制備30批大黃的供試品溶液,按“2.1”項色譜條件下進樣,記錄色譜圖。采用“中藥色譜指紋圖譜相似度評價系統”(2012版)軟件對30批大黃的HPLC圖譜進行分析。以S1號圖譜作為參照圖譜(S1號圖譜中各峰信號強度大、分離完全),設置時間窗寬度為0.1 min,通過多點校正及自動匹配生成30批大黃樣品的共有模式,采用中位數法生成對照圖譜R,并以生成的對照圖譜R為參照進行整體相似度評價。結果,共標定出了21個共有峰。以峰4(蘆薈大黃素-8-O-葡萄糖苷)作為參照峰(S)計算得到的30批大黃樣品中各共有峰相對保留時間的RSD小于1.78%(n=30),重現性較好;但其相對峰面積的RSD在31.76%~153.20%之間(n=30),波動較大,這表明不同來源大黃樣品中共有成分的含量差異較大。相似度分析結果顯示,各指紋圖譜與對照圖譜R的相似度在0.609~0.960之間,其中掌葉大黃的相似度在0.778~0.960之間,藥用大黃的相似度在0.745~0.792之間,唐古特大黃的相似度在0.609~0.690之間。30批大黃藥材指紋圖譜的共有模式、對照圖譜R見圖1,各共有峰的相對保留時間、相對峰面積分別見表2、表3,相似度分析結果見表4。
2.4.2 3種不同來源大黃對照圖譜的建立及特征成分差異峰的分析 按“2.4.1”項下指紋圖譜生成方法,分別得到20批掌葉大黃、5批藥用大黃和5批唐古特大黃樣品的指紋圖譜,并得到其相應的對照圖譜。結果顯示,3種來源大黃的對照圖譜中共有峰在數量和含量上均存在一定的差異,掌葉大黃對照圖譜中標定的共有峰為21個,藥用大黃對照圖譜中標定的共有峰為24個,而唐古特大黃對照圖譜中標定的共有峰為30個。與掌葉大黃對照圖譜相比,藥用大黃對照圖譜上增加了22~24號3個特征峰,唐古特大黃對照圖譜上增加了22號和24~31號共9個特征峰。3種不同來源大黃的對照圖譜見圖2。
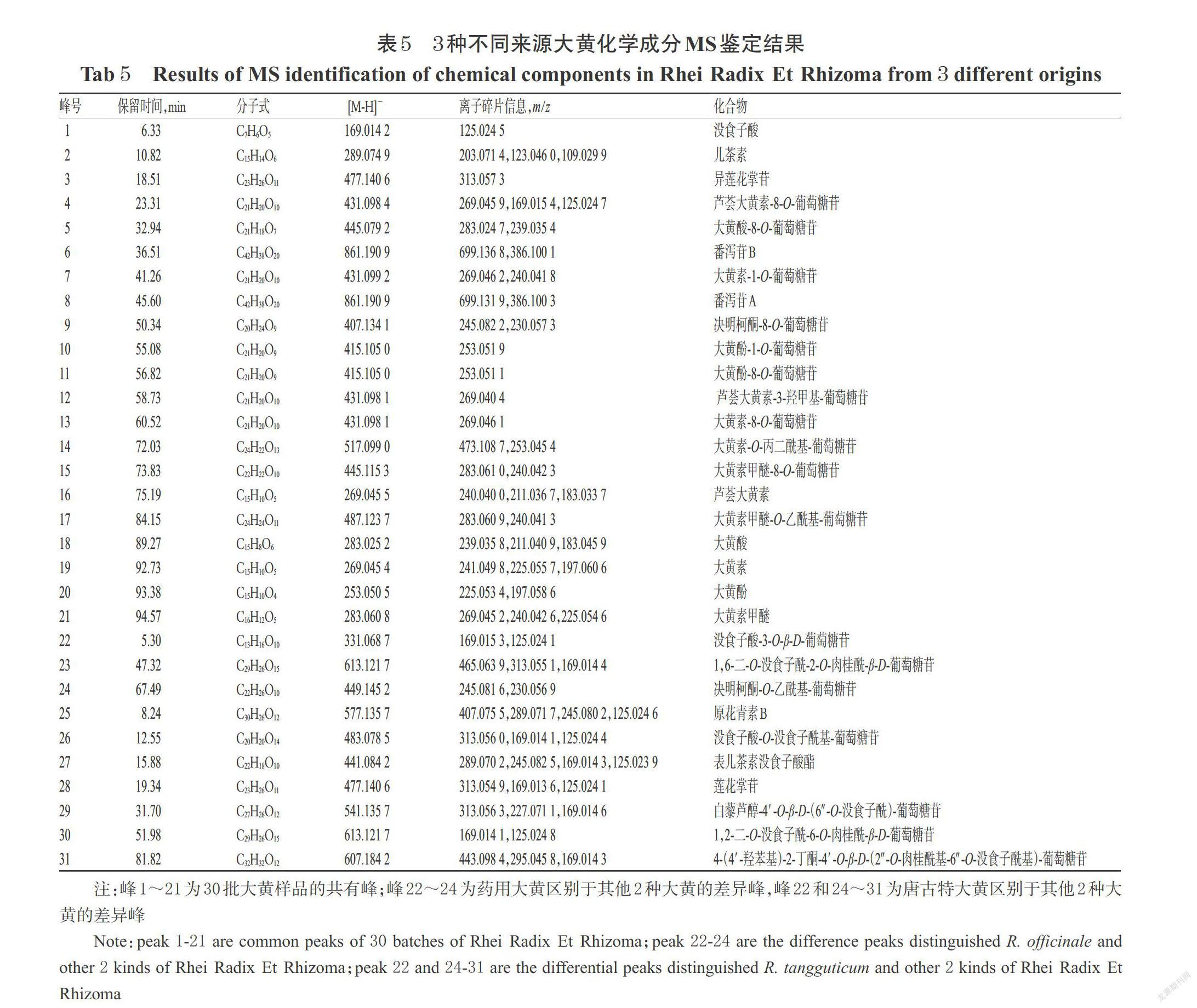
2.4.3 共有峰和特征成分差異峰的鑒定 分別按“2.1”項下方法制備3種不同來源大黃樣品(S9、S23、S28)的供試品溶液,并取“2.3”項下對照品貯備液,通過UPLC-Q-TOF-MS法對其進行檢測分析。色譜條件同“2.1”項下。MS條件為采用Dual AJS 電噴霧離子源(ESI),在負離子下模式檢測;干燥氣溫度為200 ℃,干燥氣流速為14 L/min;霧化氣壓為35 psi;鞘氣溫度為400 ℃,鞘氣流速為11 L/min;毛細管電壓為3 500 V;母離子掃描范圍為質荷比(m/z)100~1 700;二級質譜子離子掃描范圍為m/z 100~1 000;碰撞能量為-30 eV。結合對照品圖譜、樣品質譜裂解信息以及相關文獻數據[6-8],初步鑒定了21個共有成分峰和“2.4.2”中確定的3種大黃指紋圖譜之間的10個特征差異成分峰。其中,21個共有成分多為結合蒽醌、游離蒽醌及蒽酮類成分,藥用大黃區別于掌葉大黃的特征成分(22~24號峰)為鞣質和蒽酮類成分,而唐古特大黃區別于其他兩種來源大黃的特征成分(25~31號峰)包含了鞣質、苯丁酮苷和二苯乙烯類成分。3種不同來源大黃化學成分MS鑒定結果見表5。
2.4.4 PLS-DA法分析差異性成分 PLS-DA是一種集主成分分析(PCA)、典型相關分析(CCA)和多元回歸分析的基本功能為一體的具有監督模式的識別方法,可以消除眾多化學信息中相互重疊的部分,使分析數據更加準確可靠[9]。以鑒定出的30批大黃樣品的21個共有峰和10個特征差異成分峰的峰面積為變量,將其導入SIMCA-P 12.0軟件中進行PLS-DA分析,獲取PLS-DA得分圖[PLS-DA模型中,當R2Y(累積解釋能力參數)、Q2(模型預測能力參數)均大于0.5時,可認為該模型的穩定性和預測能力較好]和變量權重重要性排序(VIP)圖(當VIP>1時,表明該成分是具有統計學意義的差異標志物)[10-11]。結果顯示,建立的PLS-DA模型中R2Y和Q2分別為0.886和0.859,表明該模型穩定性及預測能力較好,可用于鑒別和區分3種來源大黃樣品。根據PLS-DA得分圖可知,大黃樣品按來源可區分為3組,能直觀顯示出各組間的差異。根據VIP圖可知,共有18個峰的VIP>1,按權重重要性排序依次為峰29>峰28>峰5>峰27>峰31>峰24>峰9>峰22>峰14>峰2>峰13>峰4>峰17>峰3>峰8>峰15>峰10>峰26,即篩選出的差異性成分分別是白藜蘆醇-4′-O-β-D-(6″-O-沒食子酰)-葡萄糖苷、蓮花掌苷、大黃酸-8-O-葡萄糖苷、表兒茶素沒食子酸酯、4-(4′-羥苯基)-2-丁酮-4′-O-β-D-(2″-O-肉桂酰基-6″-O-沒食子酰基)-葡萄糖苷、決明柯酮-O-乙酰基-葡萄糖苷、決明柯酮-8-O-葡萄糖苷、沒食子酸-3-O-β-D-葡萄糖苷、大黃素-O-丙二酰基-葡萄糖苷、兒茶素、大黃素-8-O-葡萄糖苷、蘆薈大黃素-8-O-葡萄糖苷、大黃素甲醚-O-乙酰基-葡萄糖苷、異蓮花掌苷、番瀉苷A、大黃素甲醚-8-O-葡萄糖苷、大黃酚-1-O-葡萄糖苷和沒食子酸-O-沒食子酰基-葡萄糖苷。PLS-DA得分圖和VIP得分圖分別見圖3、圖4。
3 討論
3.1 色譜條件的選擇
在前期預試驗中,筆者分別對檢測波長、流動相、柱溫等色譜條件進行了篩選:(1)檢測波長的選擇。據文獻報道[12-14],大黃中蒽醌類成分多在254、280 和430 nm波長處檢測,故本研究分別考察了在254、280、430 nm波長處大黃樣品的色譜行為,結果發現波長在254 nm時色譜峰數較多、基線最平穩、峰形較好,故本研究的檢測波長最終確定為254 nm。(2)流動相的選擇。筆者前期分別比較了以甲醇-水、乙腈-水、甲醇-0.2%甲酸水、甲醇-0.4%甲酸水為流動相體系的分離效果,結果發現以甲醇-0.4%甲酸水為流動相時,所得色譜圖基線更平穩,故本研究流動相體系選擇甲醇-0.4%甲酸水溶液。(3)柱溫的選擇。筆者前期分別考察了25、30、35 ℃柱溫對結果的影響,結果柱溫為30 ℃時峰形最佳且分離度良好,故本研究中柱溫選擇30 ℃。
3.2 樣品提取條件的考察
筆者在前期分別比較了不同提取溶劑(甲醇、75%甲醇、乙醇、75%乙醇)、不同提取方法(超聲、回流)以及不同提取時間(10、30、60 min)對樣品的提取效果。結果發現:以甲醇作為提取溶劑時提取得到的各成分含量最高;樣品經回流和超聲提取后色譜峰種類和面積均無明顯差異;在30 min提取效果最佳。為使試驗操作簡便,故選擇以甲醇為提取溶劑、超聲提取30 min的方式進行樣品處理。
3.3 大黃藥材差異成分的比較分析
據30批大黃藥材相似度分析結果顯示:僅S2、S4、S15和S19樣品的相似度達到0.9以上,其均為掌葉大黃;而藥用大黃與唐古特大黃的相似度較差,均低于0.8。通過比較3種來源大黃的對照指紋圖譜進一步發現,唐古特大黃和藥用大黃的共有峰數量較多,且鞣質類和結合蒽醌類成分顯著高于掌葉大黃(峰面積較大),而掌葉大黃中的游離蒽醌類成分含量顯著高于其他2種來源大黃(16,18~21號峰面積明顯較大)。PLS-DA分析將30批大黃分為3組,該分類趨勢可能與其來源、生長年限、生態環境具有一定的相關性,但不絕對相關。進一步通過PLS-DA模型中的VIP排序篩選出可區分不同來源大黃的18個差異性成分,這些成分可以作為快速區分不同來源大黃藥材的指標,這為大黃的質量評價以及多來源藥材的質量控制提供了參考。
參考文獻
[ 1 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2015年版.北京:中國醫藥科技出版社,2015:23-24.
[ 2 ] 桂鏡生.中藥商品學[M].昆明:云南大學出版社,2015:73-77.
[ 3 ] 逄瑜,孫磊,金紅宇,等.指紋圖譜結合化學計量學分析用于多種大黃的鑒別及質量評價[J].中國藥學雜志,2014,49(4):287-293.
[ 4 ] 杜清濤,溫金蓮,嚴優芍,等.不同品種不同產地大黃UPLC指紋圖譜研究[J].中藥材,2013,36(5):725-731.
[ 5 ] 陳安珍,蔣萬楓,袁航,等.基于超高效液相色譜偏最小二乘判別分析法建立鑒別大黃真偽及種屬預測模型的方法[J].中國藥學雜志,2016,51(3):197-202.
[ 6 ] 趙倩,陳育鵬,崔旭盛,等.掌葉大黃UPLC多指標成分測定及指紋圖譜研究[J].藥物分析雜志,2018,38(10):1697- 1710.
[ 7 ] 曹瑞,竇志華,倪麗麗,等. HPLC指紋圖譜、Q-TOF/MS定性及多成分定量相結合的大黃飲片質量評價研究[J].中草藥,2019,50(5):1100-1110.
[ 8 ] 高亮亮.唐古特大黃、藥用大黃和掌葉大黃的化學成分和生物活性研究[D].北京:北京協和醫學院,2012.
[ 9 ] 蔣萬楓,楊釗,張鳳艷.應用化學計量學和頂空-氣相色譜質譜聯用技術對7 種植物油進行類別分析[J].分析試驗室,2017,36 (6):732-737.
[10] 周欣,張琳,毛嬋,等.基于化學計量學方法結合正交偏最小二乘判別分析的陳皮飲片HPLC指紋圖譜研究[J].中草藥,2019,50(9):2194-2200.
[11] 羅益遠,劉娟秀,劉訓紅,等.基于UPLC-Triple TOF MS/MS技術分析不同產地何首烏的差異化學成分[J].質譜學報,2017,38(6):678-689.
[12] 陸文瑾,竇志華,曹瑞,等. HPLC法同時測定大黃藥材中8個非蒽醌類成分的含量[J].中國藥房,2019,30(14):1975-1980.
[13] 馮素香,王哲,郝蕊,等. HPLC法同時測定不同產地掌葉大黃中10個蒽醌類化合物[J].藥物分析雜志,2017,37(5):783-788.
[14] 竇志華,曹瑞,卞理,等.正交試驗法優選大黃中蒽醌類成分提取工藝[J].中草藥,2018,49(14):3279-3286.
(收稿日期:2019-07-18 修回日期:2019-09-06)
(編輯:林 靜)
