白芍飲片的HPLC指紋圖譜建立及聚類分析、主成分分析
2019-09-10 07:22:44林秀敏張振凌王勝超閆夢真陳祎甜張江山
中國藥房 2019年24期
林秀敏 張振凌 王勝超 閆夢真 陳祎甜 張江山









摘 要 目的:建立白芍飲片的高效液相色譜(HPLC)指紋圖譜,并進行聚類分析和主成分分析。方法:采用HPLC法。色譜柱為SunFire? C18,流動相為乙腈-0.05%磷酸水溶液(梯度洗脫),流速為1.0 mL/min,檢測波長為230 nm,柱溫為30 ℃,采集時間為70 min,進樣量為15 μL。以芍藥苷為參照,建立26批不同產地白芍飲片及30批不同炮制方法白芍飲片的HPLC指紋圖譜;采用《中藥色譜指紋圖譜相似度評價系統》(2012版)進行相似度評價,確定共有峰;采用SPSS 20.0軟件進行聚類分析和主成分分析。結果:26批不同產地白芍飲片共有9個共有峰,相似度均大于0.880;共指認了6個峰,分別為沒食子酸、兒茶素、芍藥內酯苷、芍藥苷、1,2,3,4,6-五沒食子酰葡萄糖、苯甲酰芍藥苷;聚類分析結果顯示,當余弦距離為15時26批樣品可聚為2類,S1~S21聚為一類,S22~S26聚為一類;經主成分分析,前2個主成分的累積方差貢獻率為81.124%。30批不同炮制方法白芍飲片共有10個共有峰,相似度均大于0.970;共指認了7個峰,分別為沒食子酸、兒茶素、丹皮酚新苷、芍藥內酯苷、芍藥苷、1,2,3,4,6-五沒食子酰葡萄糖、苯甲酰芍藥苷;聚類分析結果顯示,當余弦距離為25時,30批樣品可聚為2類,B1~B10聚為一類,C1~C10、J1~J10聚為一類;經主成分分析,前4個主成分的累積方差貢獻率為86.887%。結論:所建HPLC指紋圖譜及聚類分析和主成分分析結果可為不同白芍飲片的質量控制提供參考。
ABSTRACT? OBJECTIVE: To establish HPLC fingerprints of Paeonia tactilora decoction pieces, and to conduct its cluster analysis and principal component analysis. METHODS: HPLC method was adopted. The determination was performed on SunFire? C18 column with mobile phase consisted of acetonitril-0.05% phosphoric acid solution (gradient elution) at the flow rate of 1.0 mL/min. The detection wavelength was set at 230 nm, the column temperature was 30 ℃, the collection time was 70 min,and sample size was 15 μL. Using paeoniflorin as reference, HPLC fingerprints of 26 batches P. tactilora decoction pieces from different habitats and 30 batches by different processed methods were established. The similarity of samples was evaluated by TCM Chromatographic Fingerprint Similarity Evaluation System (2012 edition) to confirm common peak. Cluster analysis and principal component analysis were performed by using SPSS 20.0 software. RESULTS: There were 9 common peaks in HPLC fingerprints of 26 batches of sample from different habitats, the similarity of which was higher than 0.880. Six peaks were identified, including gallic acid, catechin, albiflorin, paeoniflorin, 1,2,3,4,6-pentagalloylglucose and benzoylpaeoniflorin. Cluster analysis showed that 26 batches of samples were clustered into 2 categories when cosine distance was 15. S1-S21 were clustered into one category; S22-S26 were clustered into the other category. By principal component analysis, the accumulative contribution rate of two main components was 81.124%. There were 10 common peaks in HPLC fingerprints of 30 batches of sample by different processed methods, the simi- larity of which was higher than 0.970. Seven peaks were identified, including gallic acid, catechin, aplopaeonoside, albiflorin, paeoniflorin, 1,2,3,4,6-pentagalloylglucose and benzoylpaeoniflorin. Cluster analysis showed that 30 batches of samples were clustered into 2 categories when cosine distance was 25. B1-B10 were clustered into one category; C1-C10 and J1-J10 were clustered into the other category. By principal component analysis, the accumulative contribution rate of four main components was 86.887%. CONCLUSIONS: Established HPLC fingerprint, the results of cluster analysis and principal component analysis can provide reference for quality control of decoction pieces of P. tactilora.
KEYWORDS? ?Paeonia tactilora decoction pieces; HPLC; Fingerprint; Cluster analysis; Principal component analysis
白芍為毛茛科植物芍藥(Paeonia lactiflora Pall.)的干燥根,其性微寒,味苦、酸,歸肝、脾經,具有養血調經、斂陰止汗、柔肝止痛、平抑肝陽之功效,可用于治療血虛萎黃、月經不調、自汗、盜汗、脅痛、腹痛、四肢攣痛、頭痛眩暈等癥[1]。現代研究表明,白芍的主要藥理作用為保肝、鎮痛、抗炎、調節免疫等[2-5]。2015年版《中國藥典》(一部)以芍藥苷作為白芍的控制指標,而赤芍的指標性控制成分也是芍藥苷,因此以芍藥苷作為白芍的質量指標,具有一定的局限性[1]。白芍中含有單萜及其苷類、三萜類、黃酮類、鞣質類和多糖類等多種化學成分[6-9],因此測定其中某個或幾個成分的含量,并不能全面反映白芍的質量。指紋圖譜以表征中藥內在質量的整體變化為目的,符合中藥質量控制整體性表征的分析特點[10-12],同時借助化學計量法進行質量評價,能更全面地體現中藥的內在品質[13-14]。為此,本研究建立了26批不同產地白芍飲片及30批不同炮制方法白芍飲片的高效液相色譜(HPLC)指紋圖譜,并結合相似度分析、聚類分析、主成分分析對其進行綜合評價,旨在為其質量控制提供參考。
1 材料
1.1 儀器
UltiMate 3000型HPLC儀,包括U3000型二極管陣列檢測器、version 7 Chromatogram 化學工作站(美國Thermo Fisher公司);600型HPLC儀,包括2487型紫外檢測器、version 2 Empower化學工作站(美國Waters公司);KQ-500 SV型超聲波清洗器(昆山市超聲儀器有限公司);BT25S型十萬分之一天平、BSA224S-CW型萬分之一天平[賽多利斯科學儀器(北京)有限公司]。
1.2 藥品與試劑
芍藥苷對照品(批號:MUST-16041901,純度:≥98.5%)、芍藥內酯苷對照品(批號:MUST-16051601,純度:≥98.5%)、1,2,3,4,6-五沒食子酰葡萄糖對照品(批號:MUST-16061211,純度:≥98.5%)、兒茶素對照品(批號:MUST-16030812,純度:≥98%)均由成都曼斯特生物科技有限公司提供;沒食子酸對照品(批號:PS-0258-0050,純度:98.5%)、苯甲酰芍藥苷對照品(批號:PS-13011802,純度:98.5%)、丹皮酚新苷對照品(批號:PS-08080102,純度:≥98%)均由成都普斯生物科技有限公司提供;乙腈、甲醇均為色譜純,磷酸為分析純,水為超純水。
1.3 藥材
白芍藥材(編號:S1~S26)經河南中醫藥大學藥學院張振凌教授鑒定為毛茛科植物芍藥(Paeonia Lactiflora Pall.)的干燥根,并按2015年版《中國藥典》(四部)[15]“炮制”通則制成飲片;白芍飲片(編號:B1~B10,中試生產)、炒白芍飲片(編號:C1~C10)、酒白芍飲片(編號:J1~J10)均由安徽亳州市滬譙藥業有限公司提供(中試生產是指按國家中藥標準化項目要求,將實驗室工藝在合作企業進行工藝放大生產,以滿足企業生產的要求)。樣品信息詳見表1。
2 方法與結果
2.1 色譜條件
色譜柱:SunFire? C18(250 mm× 4.6 mm,5 μm);流動相:乙腈(A)-0.05%磷酸水溶液(B),梯度洗脫(0~10 min,8%A→18%A;10~25 min,18%A→20%A;25~30 min,20%A;30~40 min,20%A→25%A;40~55 min,25%A →40%A;55~60 min,40%A→45%A;60~70 min,45%A→8%A);采集時間:70 min;進樣量:15 μL;流速:1.0 mL/min;柱溫:30 ℃;檢測波長:230 nm。
2.2 溶液的制備
2.2.1 混合對照品溶液 精密稱取沒食子酸對照品14.08 mg、兒茶素對照品10.90 mg、芍藥內酯苷對照品6.76 mg、芍藥苷對照品11.52 mg、丹皮酚新苷對照品10.51 mg、1,2,3,4,6-五沒食子酰葡萄糖對照品10.33 mg、苯甲酰芍藥苷對照品10.99 mg,分別置于10 mL量瓶中,加甲醇溶解并定容,得各單一對照品溶液。分別精密量取各單一對照品溶液1 mL,置于同一25 mL量瓶中,加甲醇至刻度,即得混合對照品溶液。
2.2.2 供試品溶液 精密稱取白芍中粉(中粉指能全部通過四號篩,但混有不超過60%能通過五號篩的粉末)0.1 g,置于50 mL量瓶中,加50%乙醇35 mL,超聲(功率:500 W,頻率:50 Hz)處理30 min,放冷,加50%乙醇至刻度,搖勻,經0.22 μm微孔濾膜濾過,取續濾液,即得。
2.3 方法學考察
2.3.1 精密度試驗 取“2.2.2”項下供試品溶液(編號:S10)適量,按“2.1”項下色譜條件連續進樣測定6次,以芍藥苷峰的保留時間和峰面積為參照,記錄各共有峰的相對保留時間和相對峰面積。結果,9個共有峰相對保留時間的RSD均小于0.19%(n=6),相對峰面積的RSD均小于2.34%(n= 6),表明本方法精密度良好。
2.3.2 穩定性試驗 取“2.2.2”項下供試品溶液(編號:S10)適量,分別于室溫下放置0、2、4、8、12、24 h時按“2.1”項下色譜條件進樣測定,以芍藥苷峰的保留時間和峰面積為參照,記錄各共有峰的相對保留時間和相對峰面積。結果,9個共有峰相對保留時間的RSD均小于0.21%(n=6),相對峰面積的RSD均小于2.61%(n=6),表明供試品溶液于室溫下放置24 h內穩定性良好。
2.3.3 重復性試驗 精密稱取樣品(編號:S10)0.1 g,共6份,按“2.2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,以芍藥苷峰的保留時間和峰面積為參照,記錄各共有峰的相對保留時間和相對峰面積。結果,9個共有峰相對保留時間的RSD均小于0.34%(n=6),相對峰面積的RSD均小于2.75%(n= 6),表明本方法重復性良好。
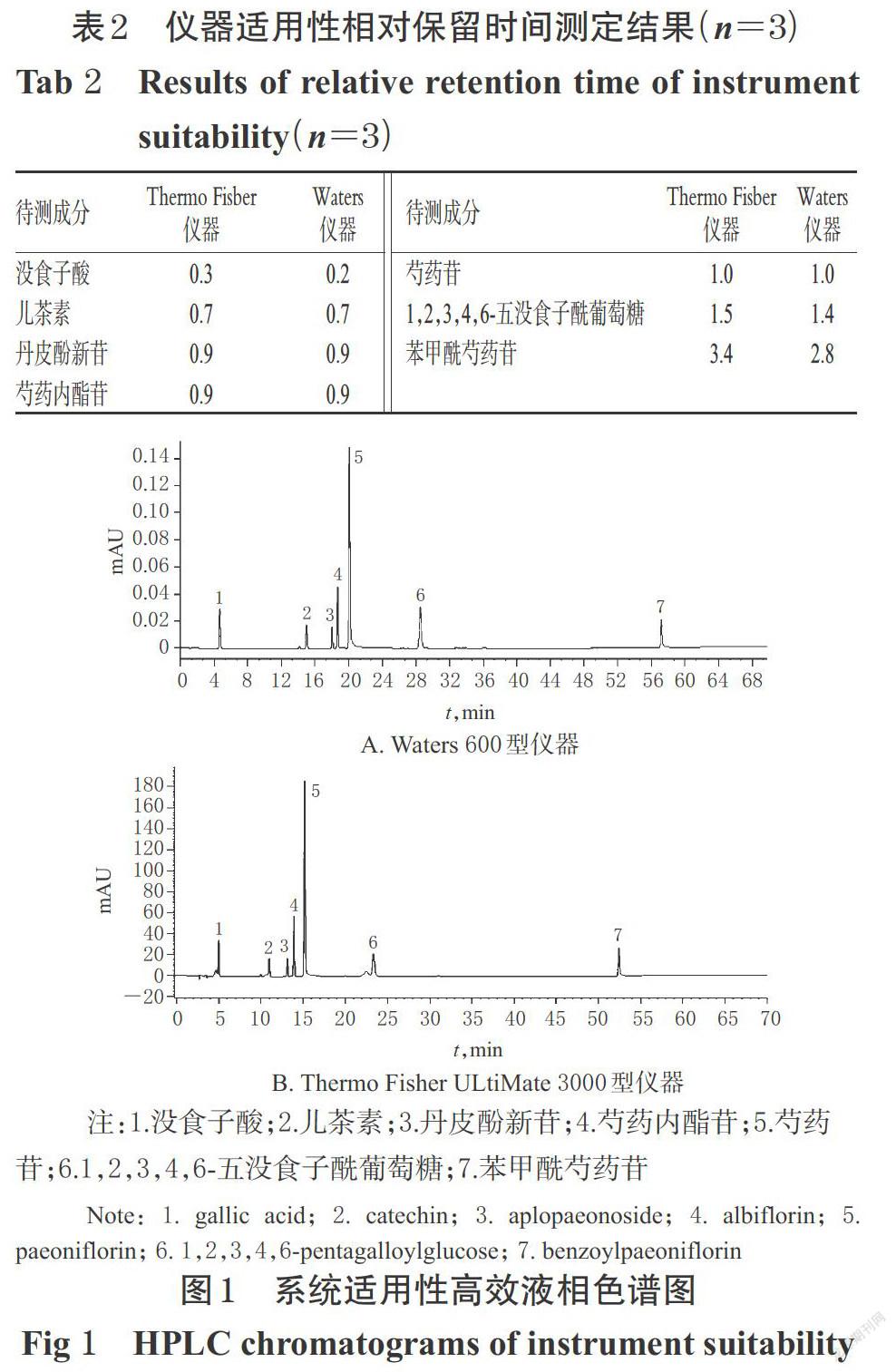
2.3.4 儀器適用性試驗 取“2.2.1”項下混合對照品溶液5 μL,按“2.1”項下色譜條件進樣測定,以芍藥苷為參照,計算沒食子酸、兒茶素、丹皮酚新苷、芍藥內酯苷、芍藥苷、1,2,3,4,6-五沒食子酰葡萄糖和苯甲酰芍藥苷的相對保留時間。通過配對t檢驗,考察不同儀器對色譜條件的適用性。結果,t=1.38,雙側P=0.22>0.05,表明相同色譜條件下,以芍藥苷為參照,兩種儀器所測色譜峰可定位其他成分的相對保留時間,且無顯著性差異,不同儀器不影響目標峰的出峰順序,相對定位準確,詳見表2、圖1。
2.4 不同產地白芍飲片HPLC指紋圖譜的生成與相似度、共有峰分析
2.4.1 HPLC指紋圖譜的生成 取26批不同產地白芍飲片(編號:S1~S26)各0.1 g,按“2.2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,采用《中藥色譜指紋圖譜相似度評價系統》(2012版)進行分析,得HPLC指紋圖譜,詳見圖2、圖3。
2.4.2 相似度評價 采用《中藥色譜指紋圖譜相似度評價系統》(2012版),以樣品的HPLC對照指紋圖譜為對照,進行整體相似度評價。結果,26批不同產地白芍飲片(編號:S1~S26)的相似度為0.887~0.998,提示不同產地白芍存在微小的質量差異,詳見表3。
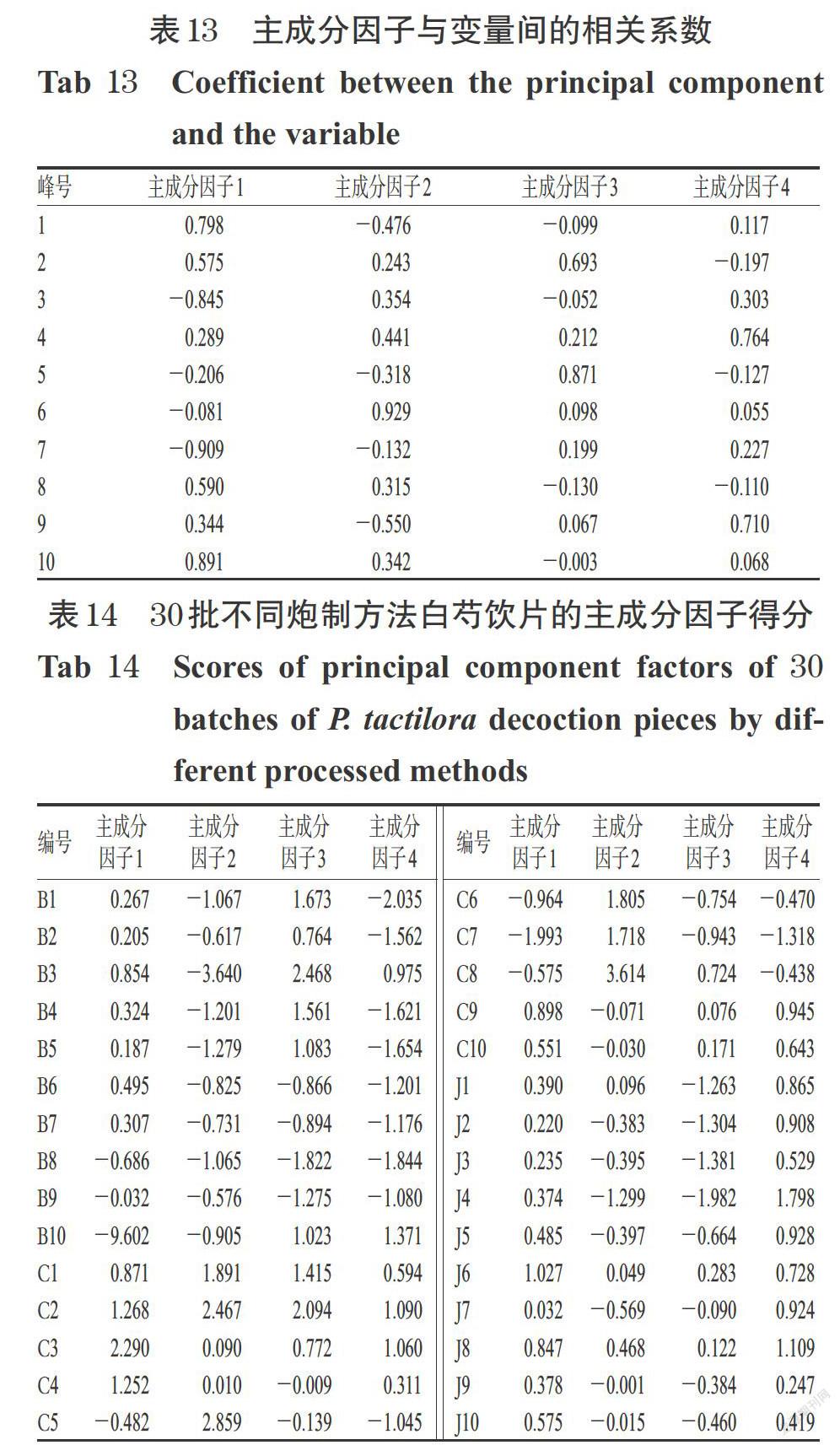
2.4.3 共有峰的指認及相關分析 26批不同產地白芍飲片(編號:S1~S26)共有9個共有峰,通過與對照品圖譜比對,共指認了6個色譜峰,分別為沒食子酸(峰2)、兒茶素(峰3)、芍藥內酯苷(峰4)、芍藥苷(峰5)、1,2,3,4,6-五沒食子酰葡萄糖(峰7)、苯甲酰芍藥苷(峰9)。因芍藥苷的峰面積較大、分離度較好,故以其為參照峰,計算其他成分的相對保留時間和相對峰面積,詳見表4、表5。
2.4.4 不同產地白芍飲片鏡像對比 隨機選取3批不同產地白芍飲片(編號:S9、S16、S22)的圖譜進行鏡像對比,詳見圖4。由圖4可知,在同一檢測條件下,亳白芍(S9)檢測出1個色譜峰,經與對照品比對判定為丹皮酚新苷,而杭白芍(S22)、川白芍(S16)未檢測出該成分。
2.4.5 聚類分析 以各共有峰的峰面積除以稱樣量的數值作為原始數據,采用SPSS 20.0 軟件,以組間連接法結合余弦距離對26批不同產地白芍飲片(編號:S1~S26)進行聚類分析,詳見圖5。由圖5可知,當余弦距離為15時,樣品可聚為2類,S1~S21聚為一類,S22~S26聚為一類;當余弦距離小于5時,樣品可聚為5類,S1~S15聚為一類,S16~S21聚為一類,S22、S23聚為一類,S24、S26聚為一類,S25聚為一類。
2.4.6 主成分分析 對26批不同產地白芍飲片的9個共有峰的絕對峰面積進行標準化處理后,采用SPSS 20.0 軟件進行主成分分析。結果顯示,共得到2個主成分因子,特征值分別為6.002、1.299,方差貢獻率分別為66.686%、14.438%,累積方差貢獻率為81.124%,提示主成分因子1、2可作為不同產地白芍飲片的評價指標。故以主成分因子1、2為指標對26批不同產地白芍飲片(S1~S26)進行評價。結果顯示,主成分因子1與9個共有峰相關性均較強,特別是峰1、峰7和峰8;主成分因子2對峰5、峰6的影響較大;主成分因子1得分對S16~S26影響較大,主成分因子2對S1~S15 影響較大,以峰面積為指標進行歸一化,結果顯示亳白芍、川白芍、杭白芍之間存在差異,詳見表6~表8。
2.5 不同炮制方法白芍飲片HPLC指紋圖譜的生成與相似度、共有峰分析
2.5.1 不同炮制方法白芍飲片HPLC指紋圖譜的生成 按“2.2.2”項下方法分別制備白芍(編號:B1~B10)、炒白芍(編號:C1~C10)、酒白芍(編號:J1~J10)供試品溶液,按“2.1”項下色譜條件進樣測定,采用《中藥色譜指紋圖譜相似度評價系統》(2012版)進行分析,得到HPLC指紋圖譜,詳見圖6、圖7。
2.5.2 相似度評價 采用《中藥色譜指紋圖譜相似度評系統》(2012版),以樣品的HPLC對照指紋圖譜為對照,進行整體相似度評價。結果,30批不同炮制方法白芍飲片與對照指紋圖譜之間差別不明顯,相似度均大于0.970,表明不同炮制方法白芍飲片間差異不大,詳見表9。
2.5.3 共有峰的指認及相關分析 30批不同炮制方法白芍飲片共有10個共有峰,通過與對照品比對,共指認了7個色譜峰,分別為沒食子酸(峰1)、兒茶素(峰2)、丹皮酚新苷(峰3)、芍藥內酯苷(峰4)、芍藥苷(峰5)、1,2,3,4,6-五沒食子酰葡萄糖(峰8)、苯甲酰芍藥苷(峰10)。因芍藥苷的峰面穩定、分離度良好,故以其為參照峰,計算其他成分的相對保留時間和相對峰面積,詳見表10、表11。
2.5.4 不同白芍炮制飲片鏡像對比 取白芍飲片(編號:B1)、炒白芍飲片(編號:C1)、酒白芍飲片(編號:J1)的圖譜進行鏡像對比,詳見圖8。由圖8可知,在同一檢測條件下,酒白芍檢測到1個未知峰,白芍、炒白芍均未檢出該成分。
2.5.5 聚類分析 按“2.4.5”項下方法對30批不同白芍炮制飲片(編號:B1~B10、C1~C10、J1~J10)進行聚類分析,詳見圖9。由圖9可知,當余弦距離為25時,樣品可聚為2類,B1~B10聚為一類,C1~C10、J1~J10聚為一類;當余弦距離為20時,可聚為4類,B1~B10聚為一類,C5~C8聚為一類,C1~C4、C9、C10聚為一類,J1~J10聚為一類。
2.5.6 主成分分析 對30批不同炮制方法白芍飲片的10個共有峰的絕對峰面積進行標準化處理后,采用SPSS 20.0 軟件進行主成分分析。結果顯示,共得到4個主成分因子,特征值分別為3.899、2.105、1.365、1.319,方差貢獻率分別為38.988%、21.052%、13.654%、13.193%,累積方差貢獻率為86.887%,提示主成分因子1、2、3、4可作為不同炮制方法白芍飲片的評價指標。故以主成分因子1、2、3、4為指標對30批不同炮制方法白芍飲片(B1~B10、C1~C10、J1~J10)進行評價。結果顯示,主成分因子1對峰1和峰10影響較大,主成分因子2對峰6影響較大,主成分因子3對峰5影響較大,主成分因子4對峰4和峰9影響較大;主成分因子1和主成分因子4得分在樣品J1~J10中較高,表明其對酒白芍影響較顯著;主成分因子2得分對樣品C1~C10影響較大,表明其對炒白芍影響較明顯;主成分因子3得分對樣品B1~B10影響較大,表明其對白芍飲片影響突出。以10個共有峰的峰面積進行歸一化,結果顯示不同炮制方法白芍飲片存在質量差異,詳見表12~表14。
3 討論
本研究前期對液相條件進行了優化,發現以乙腈-0.05%磷酸水溶液為流動相進行梯度洗脫時,各成分的分離效果較好,保留時間適當,峰形良好。同時,采用二極管陣列檢測器在190~400 nm波長內進行掃描,得三維光譜圖。結果顯示,檢測波長為214 nm時,1,2,3,4,6-五沒食子酰葡萄糖、沒食子酸峰有最大吸收,但峰形較差,基線波動較大,對樣品峰面積的積分影響大;當檢測波長為230 nm時,芍藥苷峰有最大吸收,且色譜峰形較好,出峰數目較多,各色譜峰之間分離度較好,基線平穩,故選擇230 nm為檢測波長。
26批不同產地白芍飲片的整體相似度較高,共有化學成分沒食子酸、芍藥苷、1,2,3,4,6-五沒食子酰葡萄糖、苯甲酰芍藥苷等較穩定,提示不同產地間白芍飲片的化學成分接近;但亳白芍比杭白芍、川白芍多了1個丹皮酚新苷峰,后續將擴大樣本量進一步研究。26批不同產地白芍飲片共有9個共有峰,其相對保留時間的RSD為0~0.97%,相對峰面積的RSD為0~74.45%,表明不同產地白芍飲片中雖然化學成分相似,但含量存在差異。聚類分析和主成分分析結果顯示,當余弦距離為15時,26批不同產地白芍飲片可聚為2類,S1~S21聚為一類,S22~226聚為一類,提示不同產地白芍飲片的成分含量存在差異,可能與生長環境不同有關[16]。
30批不同炮制方法白芍飲片的整體相似度較高,但酒白芍檢測出1個未知峰,炒白芍和白芍飲片未檢出。30批不同炮制方法白芍飲片共有10個共有峰,其相對保留時間的RSD為0~0.59%,相對峰面積的RSD為0~92.89%,表明白芍經不同方法炮制后,其化學成分含量變化較大[17-18]。聚類分析和主成分分析結果顯示,當余弦距離為25時,30批不同炮制方法白芍飲片可聚為2類,B1~B10聚為一類,C1~C10、J1~J10聚為一類,提示白芍炮制飲片具有明顯的分類趨勢,間接證明其經炮制后內部化學成分可能發生變化,體現出不同炮制飲片的差異性[17]。
綜上所述,本研究建立的HPLC指紋圖譜及聚類分析和主成分分析結果可為不同白芍飲片的質量控制提供參考。
參考文獻
[ 1 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2015年版.北京:中國醫藥科技出版社,2015:105.
[ 2 ] 章麗,趙冰潔,袁嘉瑞,等.牡丹皮、赤芍與白芍對急性血瘀模型大鼠活血功效的比較研究[J].中草藥,2016,47(15):2676-2683.
[ 3 ] 牟翔宇,郭英慧,孫文君,等.柴胡配伍白芍治療PMDD肝氣郁證的研究進展[J].中國實驗方劑學雜志,2018,24(20):192-199.
[ 4 ] 左志燕,詹淑玉,黃嬛,等.白芍總苷保肝作用的藥動學和藥效學研究進展[J].中國中藥雜志,2017,42(20):3860- 3865.
[ 5 ] 王成龍.基于白芍養血柔肝功效的芍藥苷、芍藥內酯苷藥理作用研究[D].北京:北京中醫藥大學,2017.
[ 6 ] 笪婧雯.白芍的化學成分研究[J].海峽藥學,2017,29(12):45-46.
[ 7 ] 金林,趙萬順,郭巧生,等.白芍飲片的化學成分測定及質量評價[J].中國中藥雜志,2015,40(3):484-489.
[ 8 ] 范瑪莉,邢婕,李震宇,等.基于NMR代謝組學技術的白芍與赤芍化學成分比較研究[J].中草藥,2014,45(22):3230-3237.
[ 9 ] 周海玲,許舜軍,周若龍,等.白芍、赤芍化學成分的高效液相色譜-飛行時間串聯質譜分析[J].中藥材,2018,41(7):1637-1640.
[10] 劉東方,趙麗娜,李銀峰,等.中藥指紋圖譜技術的研究進展及應用[J].中草藥,2016,47(22):4085-4094.
[11] 鄒純才,鄢海燕.我國中藥色譜指紋圖譜相似度評價方法30年(1988-2017年)研究進展與展望[J].中國中藥雜志,2018,43(10):1969-1977.
[12] 張小藝,劉久石,高石曼,等.中藥譜效關系的研究方法及應用進展[J/OL]. [2019-08-27]. https://doi.org/10.19540/-
j.cnki.cjcmm.20190429.201.
[13] 黃華花,王明軍,黃鳴清,等.金橘藥材的UPLC指紋圖譜建立、聚類分析及主成分分析[J].中國藥房,2019,30(12):1661-1665.
[14] 林林,于鳳蕊,徐麗華,等.女金丸HPLC指紋圖譜建立及聚類分析和主成分分析[J].中國藥房,2019,30(10):1339- 1343.
[15] 國家藥典委員會.中華人民共和國藥典:四部[S]. 2015年版.北京:中國醫藥科技出版社,2015:31.
[16] 童黃錦,白發平,汪小莉,等.不同產地白芍藥材的指紋圖譜研究[J].中醫學報,2014,29(9):1326-1329.
[17] 萬超,蘇慧,霍雨佳,等.白芍飲片炒制前后物質基礎的變化規律分析[J].中國實驗方劑學雜志,2018,24(22):28- 33.
[18] 任娟,劉曉,李偉東,等.基于UHPLC-Q-TOF-MS/MS的白芍炒制前后化學成分研究[J].世界中醫藥,2019,14(2):268-273.
(收稿日期:2019-06-22 修回日期:2019-10-29)
(編輯:陳 宏)
