高效液相色譜法測定伏格列波糖口腔速溶膜中伏格列波糖的含量和有關物質
2019-10-16 08:19:18孫文霞成都大學四川抗菌素工業研究所四川成都610052
成都大學學報(自然科學版) 2019年3期
孫文霞, 陳 勇(成都大學 四川抗菌素工業研究所, 四川 成都 610052)
0 引 言
伏格列波糖,化學名為(+)-1L-[1(羥基),2,4,5/3]-5-[2-羥基-1-(羥甲基)乙基]氨基-1-碳-(羥甲基)-1,2,3,4-環已四醇,是一種從放線菌培養液中發現的天然糖類似物,能夠抑制小腸壁細胞a-葡萄糖苷酶的活性,從而有效改善糖尿病餐后血糖,目前廣泛用于I、II型糖尿病的防治[1-5].2006年,伏格列波糖口腔速溶膜劑面世,并得到廣泛使用,但目前關于控制其質量的相關文獻較少.對此,本研究通過建立柱后衍生HPLC熒光檢測法,測定伏格列波糖口腔速溶膜中伏格列波糖的含量和有關物質,以有效控制伏格列波糖口腔速溶膜劑的質量.
1 材料與儀器
1.1 材 料
實驗所用材料包括:伏格列波糖對照品(批號,100826-200801,純度99.2%),由中國食品藥品檢定研究院提供;伏格列波糖口腔速溶膜(批號,140711、140712、140713,規格為0.2 mg),由四川抗菌素工業研究所提供;伏格列波糖口腔速溶膜(批號,4C11W,規格為0.2 mg),購自日本救急藥品工業株式會社;乙腈為色譜純,購自美國fisher公司;水為重蒸餾水,其余試劑均為分析純.
1.2 儀 器
實驗所用儀器包括:LC-20AB型高效液相色譜儀、RF-10AXL型熒光檢測器、CRB-6A型化學反應箱、LC-Solution色譜工作站(日本島津公司);FE-20型pH計(梅特勒—托利多公司);CPA-225D型十萬分之一電子天平(賽多利斯科學儀器(北京)有限公司).
2 方法和結果
2.1 色譜條件
實驗的色譜條件為:含量測定用色譜柱為COSMOSIL 5C18-PAQ柱(250 mm×4.6 mm,10 μm);有關物質測定用色譜柱為Shodex Asahipak NH2P-50 4E柱(250 mm×4.6 mm,5 μm);含量測定流動相為0.045%辛烷磺酸鈉的磷酸鹽緩沖液(取3.12 g磷酸二氫鈉二水合物,加水1 000 mL使其溶解,用磷酸調pH值至3.5)—甲醇(94∶6);有關物質測定流動相為磷酸緩沖液(取1.56 g磷酸二氫鈉二水合物溶解于500 mL水中,另取3.58 g十二水磷酸氫二鈉溶解于500 mL水中,用磷酸調pH值至6.5, 將2種溶液混合均勻)—乙腈(37∶63);熒光檢測器激發波長為350 nm,發射波長為430 nm;柱溫為25 ℃;熒光衍生試劑的制備方法為,取牛磺酸6.25 g、高碘酸鈉2.56 g溶解至1 000 mL水中,搖勻,即得;反應浴溫度為100 ℃,聚四氟乙烯反應管長20 m(內徑0.5 mm),冷卻浴溫度為15 ℃,聚四氟乙烯冷卻管長2 m(內徑0.3 mm);流動相與熒光試劑流速相同,含量測定為1.0 mL/min,有關物質測定為0.6 mL/min;進樣量100 μL.
2.2 溶液配制
2.2.1 供試品溶液配制.
取本品10片,剪碎,置于50 mL量瓶中,加流動相40 mL,超聲處理10 min,放冷,用流動相稀釋至刻度,搖勻,過濾,精密量取續濾液1 mL,置10 mL量瓶中,用流動相稀釋至刻度,搖勻,作為含量測定用供試品液.另取本品10片,剪碎,置5 mL量瓶中,加流動相4 mL,超聲處理10 min,放冷,用流動相稀釋至刻度,搖勻,過濾,取續濾液作為有關物質檢查用供試品溶液.
2.2.2 對照品溶液配制.
精密稱取伏格列波糖對照品10 mg,置25 mL量瓶中,用流動相溶解并稀釋至刻度,搖勻,精密量取1 mL,置10 mL量瓶中,用流動相稀釋至刻度,搖勻,作為對照品溶液.
2.2.3 對照溶液配制.
精密量取1.0 mL“2.2.1”項下有關物質檢查用供試品溶液,置100 mL量瓶中,用流動相稀釋至刻度,搖勻,作為有關物質檢查用對照溶液.
2.3 含量測定
精密量取“2.2”項下含量測定用供試品溶液和對照品溶液各100 μL,注入液相色譜儀,記錄色譜圖.按外標法計算出供試品中伏格列波糖的含量.
2.4 有關物質測定
精密量取“2.2”項下對照溶液100 μL注入液相色譜儀,調節檢測靈敏度,使主成分色譜峰的峰高為滿量程的20%.再精密量取“2.2”項下有關物質檢查用供試品溶液100 μL注入液相色譜儀,記錄色譜圖至主成分峰保留時間的2.5倍.供試品溶液的色譜圖中如顯雜質峰,扣除輔料峰后,單個雜質峰面積不得大于對照溶液峰面積的0.2倍(0.2%),各個雜質峰面積之和不得大于對照溶液峰面積的0.5倍(0.5%).
2.5 方法驗證
2.5.1 專屬性.
取本品適量(批號140711,約相當于伏格列波糖2.0 mg),置于5 mL的容量瓶中,平行制備6份,進行破壞性實驗,具體包括:
1)高溫破壞.樣品經100 ℃高溫加熱約4 h;
2)光照破壞.樣品在光照度為(4 500±500) lx下放置10 d.
3)氧化破壞.樣品加30%雙氧水0.5 mL,放置1 h.
4)酸破壞.樣品中加1 mol/L的鹽酸0.5 mL,放置4 h后,用1 mol/L的氫氧化鈉至中性.
5)堿破壞.樣品加1 mol/L的NaOH溶液0.5 mL,放置4 h,加1 mol/L 的HCl溶液至中性.
上述經破壞的樣品均用流動相稀釋至刻度,搖勻,過濾,取續濾液按照“2.1”項下色譜條件進行測定.結果顯示:本品經光照和高溫破壞,主峰面積有所下降,雜質個數和峰面積均有所增加;在經酸、堿和雙氧水破壞后主峰面積明顯降低,說明該藥對酸、堿和雙氧水不穩定.相鄰雜質與主成分色譜峰分離較好,分離度大于1.5,說明所建方法的專屬性強.破壞性實驗色譜如圖1所示.

圖1 破壞性實驗色譜圖
2.5.2 線性關系.
精密稱取伏格列波糖對照品適量,用流動相制成每1 mL中含伏格列波糖25.0、32.0、40.0、45.0、50.0 μg的溶液,分別精密吸取100 μL注入液相色譜儀,記錄色譜圖.以峰面積Y對伏格列波糖X(μg/mL)進行線性回歸,得回歸方程為,Y=243 345X-811 215.73(R=0.9999,n=5).結果表明,伏格列波糖濃度與色譜峰面積在25.0~50.0 μg/mL范圍內線性關系良好.
2.5.3 進樣精密度.
精密量取“2.2”項下對照品溶液和對照溶液各100 μL,分別注入液相色譜儀,記錄色譜圖,連續測定6次,峰面積的RSD分別為0.52%和0.76%.結果表明,儀器的進樣精密度良好.
2.5.4 重復性.
取同一批樣品(批號,140711),按“2.2.1”項下方法平行制備6份含量測定用供試品液,按“2.1”項下色譜條件分別進樣分析,計算得到含量分別為100.23%、100.06%、100.35%、100.74%、101.45%和101.63%,RSD為0.65%(n=6).結果表明,方法的重復性良好.
2.5.5 中間精密度.
精密稱取樣品(批號,140711),按照“2.2”項下的方法配制成含量測定用供試品溶液和對照品溶液,于不同日期,不同操作人員在不同儀器上按“2.3”項下方法測定,并按外標法計算伏格列波糖口腔速溶膜的含量,其含量的RSD為0.77%(n=6).結果表明,中間精密度良好.
2.5.6 回收率.
取本品10片置100 mL容量瓶中,加流動相80 mL,照“2.2.1”項下方法制成濃度為0.02 mg/mL的供試品母液,精密量取該母液1 mL置10 mL容量瓶中,平行制備9份,分別加入“2.2.2”項下對照品溶液4 mL(80%)、5 mL(100%)、6 mL(120%),用流動相稀釋至刻度,每個濃度平行制備3份.按“2.3”項下方法測定,計算回收率,結果如表1所示.
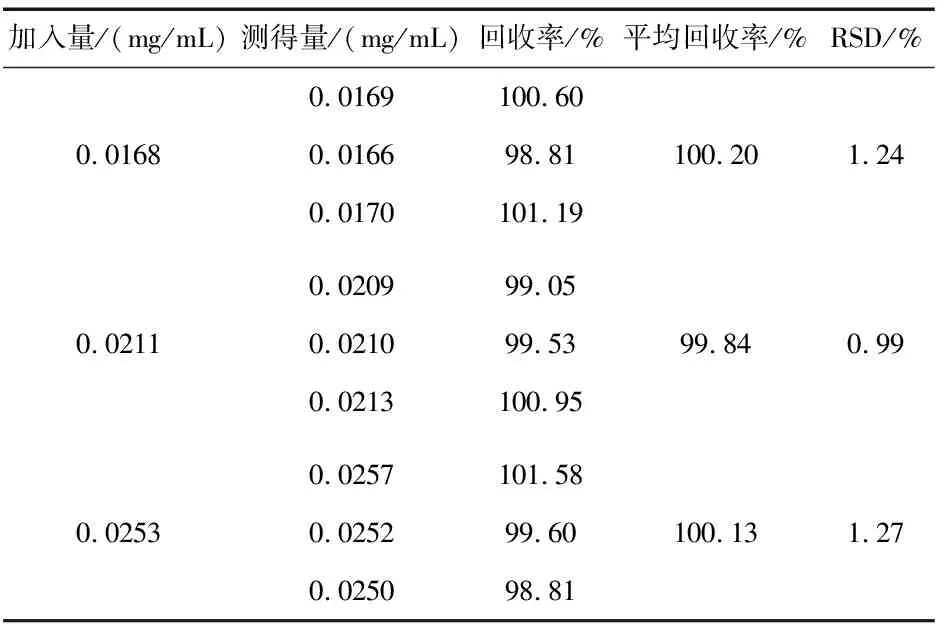
表1 回收率實驗結果
2.5.7 檢測限與定量限.
取“2.2.2”項下對照品溶液逐級稀釋,按“2.1”項下色譜條件測定,以信噪比S/N=3和S/N=10計,測得伏格列波糖的檢測限和定量限分別為2 ng和10 ng.
2.6 樣品測定結果
取3批樣品,照“2.3”和“2.4”項下方法進行含量和有關物質測定,結果見表2.

表2 樣品含量和有關物質檢測結果
3 討 論
由于伏格列波糖分子結構中不含共軛基團,無紫外吸收,因而無法采用HPLC-UV法.目前相關文獻中測定伏格列波糖的方法主要有HPLC-ELSD法[6]、HPLC-RID法[7]、離子色譜法[8]、柱前衍生化GC法[9]、柱后衍生HPLC熒光檢測法[10-14]以及高效液相色譜—質譜法[15].伏格列波糖口腔速溶膜劑作為一種新的劑型,尚未有關于其有關物質和含量測定的相關文獻報道.對此,本研究參考日本藥典中伏格列波糖原料和伏格列波糖片劑的標準,采取柱后衍生HPLC熒光檢測法進行相關物質與含量的測定.
在日本的藥典中,采用氨基丙基硅烷填料色譜柱進行含量測定,考慮到該色譜柱用量較少,結合本實驗室的條件,參考國家藥品標準,采用C18色譜柱.由于長時間運行純水相流動相時柱子使用壽命較短,故對流動相條件進行優化,當流動相為0.045%辛烷磺酸鈉的磷酸鹽緩沖液(取3.12 g磷酸二氫鈉二水合物,加水1 000 mL使溶解,用磷酸調pH值至3.5)—甲醇(94∶6),伏格列波糖的保留時間以及對稱性較好,但是考慮到水相比例仍較高,因而選擇親水性很好的COSMOSIL 5C18-PAQ柱.此外,在日本的藥典中,伏格列波糖原料藥對3個已知雜質即雜質1、雜質2及雜質3分別進行了限定.而文獻[16]的研究表明,該3個雜質為原料藥合成工藝過程所引入,并非伏格列波糖降解雜質.結合本研究的破壞實驗結果,其均未在相應保留時間處產生雜質峰,故本研究認為可不對本品中3個已知雜質進行限制.
