超臨界流體萃取與模擬移動(dòng)床色譜純化靈芝三萜類(lèi)化合物
2019-10-29 06:38:52余書(shū)奇包曉青梁明在金晨鐘
食品科學(xué) 2019年20期
關(guān)鍵詞:實(shí)驗(yàn)
余書(shū)奇,包曉青,梁明在*,金晨鐘,田 蔚
(1.湖南人文科技學(xué)院農(nóng)業(yè)與生物技術(shù)學(xué)院,湖南 婁底 417000;2.璞勵(lì)卓越(北京)科技有限公司,北京 100000;3.義守大學(xué)化工系,中國(guó)臺(tái)灣 高雄 84001)
靈芝作為傳統(tǒng)的中藥材具有很高的藥用價(jià)值,已經(jīng)成為保健食品的主要原料之一。研究發(fā)現(xiàn)靈芝具有調(diào)節(jié)免疫系統(tǒng)、心血管系統(tǒng)等藥理功能[1],其中靈芝三萜類(lèi)化合物是靈芝的關(guān)鍵藥效成分之一,主要指標(biāo)成分為靈芝酸A、靈芝酸F和靈芝醇B,現(xiàn)代藥理學(xué)研究表明靈芝三萜類(lèi)化合物具有抗腫瘤、保肝護(hù)肝、抗菌消炎等功能[2-6]。因此,對(duì)靈芝中的靈芝三萜類(lèi)化合物進(jìn)行有效提取及純化,可拓寬靈芝產(chǎn)品多元化發(fā)展。目前,靈芝三萜類(lèi)化合物的提取多采用有機(jī)溶劑提取、超聲波輔助提取等方式。而采用超臨界二氧化碳提取可提高靈芝三萜類(lèi)化合物的穩(wěn)定性,并且綠色無(wú)污染,安全無(wú)毒[7-9]。研究顯示超臨界流體萃取的操作條件對(duì)產(chǎn)物中靈芝三萜類(lèi)化合物的組分種類(lèi)和活性有一定程度的影響[10-13],因此本實(shí)驗(yàn)優(yōu)化了超臨界流體萃取的最佳條件與方法;同時(shí)采用模擬移動(dòng)床(simulated moving bed,SMB)移除粗萃物中的不純物以提高靈芝三萜類(lèi)化合物含量。SMB技術(shù)是一種現(xiàn)代化精細(xì)的色譜分離技術(shù),在固定床和真實(shí)移動(dòng)床的基礎(chǔ)上發(fā)展而來(lái)[14-15]。該技術(shù)采用固定相與移動(dòng)相連續(xù)式地逆向流動(dòng),大幅提高了固定相的使用效率,達(dá)到連續(xù)進(jìn)料提高產(chǎn)量的目的[15-17]。由于其相對(duì)于傳統(tǒng)制備色譜分離技術(shù)具有能連續(xù)化操作、易實(shí)現(xiàn)自動(dòng)化、分離能力強(qiáng)、分離效率高等特點(diǎn),因而在食品科學(xué)和生物化學(xué)、醫(yī)學(xué)領(lǐng)域得到了越來(lái)越廣泛的應(yīng)用[18-22],目前,食品領(lǐng)域中SMB的應(yīng)用熱點(diǎn)在天然產(chǎn)物的提取純化上,如鐵皮石斛[23]、白藜蘆醇[24]、紫杉醇[25]、甜葉菊苷[26]、甘草苷[27]等的純化,曾有研究者利用使用超臨界流體的SMB進(jìn)行了中國(guó)臺(tái)灣牛樟芝三萜的分離純化研究[28],但國(guó)內(nèi)外尚未有SMB純化靈芝三萜類(lèi)化合物的應(yīng)用報(bào)道。
1 材料與方法
1.1 材料與試劑
靈芝子實(shí)體由浙江壽仙谷公司提供,為赤靈芝。
靈芝酸A(>98%)、靈芝酸F(>98%)、靈芝醇B(>98%)標(biāo)準(zhǔn)品 成都曼思特股份有限公司;95%乙醇景明化工股份有限公司;乙腈(色譜純) 友和貿(mào)易股份有限公司;二氧化碳30 kg(插管、高純度)錦德氣體股份有限公司。
1.2 儀器與設(shè)備
高效液相色譜( high performance liquid chromatography,HPLC,泵:2130,紫外檢測(cè)器:L-2455)儀 日本Hitachi公司;超臨界萃取設(shè)備(萃取槽體積為1 L,10.5 cm×11.1 cm) 中國(guó)臺(tái)灣金屬工業(yè)研究中心;SMB色譜(1/8不銹鋼配管,配置8 支管柱,1.0 cm×25 cm) 中國(guó)臺(tái)灣喬璞科技有限公司。
1.3 方法
1.3.1 超臨界流體萃取條件優(yōu)化
取1 3 7 g的靈芝子實(shí)體切粒(顆粒直徑約1~3 mm),置于萃取槽中,在350 bar、45 ℃進(jìn)行萃取,每0.5 h從分離槽中取樣一次,分離槽溫度設(shè)定為50 ℃,壓力設(shè)定為45 bar,持續(xù)萃取6 h,根據(jù)萃取液中目標(biāo)物含量的變化確定萃取時(shí)間。考察夾帶劑對(duì)萃取結(jié)果的影響:A組不添加夾帶劑,二氧化碳流速為60 g/min,在萃取過(guò)程中同時(shí)從萃取槽出口端以5 mL/min流速泵入乙醇溶液,以避免萃取物阻塞;B組采用乙醇為夾帶劑,流速為5 mL/min,二氧化碳流速為60 g/min,在進(jìn)入萃取槽前便與二氧化碳混合后再加熱,共同進(jìn)入萃取槽中萃取。
1.3.2 萃取液中靈芝三萜類(lèi)化合物含量的測(cè)定
采用1.3.1節(jié)B組的方式對(duì)靈芝進(jìn)行萃取,所得萃取液用HPLC測(cè)定目標(biāo)組分含量。分析方法參照文獻(xiàn)[29]:色譜柱:Agilent Eclipse XDB-C18(250 mm×4.6 mm,5 μm),流動(dòng)相按表1進(jìn)行梯度洗脫,分析波長(zhǎng)252 nm,流速1 mL/min,進(jìn)樣量20 μL。繪制靈芝酸A、靈芝醇B及靈芝酸F標(biāo)準(zhǔn)曲線(xiàn),所得曲線(xiàn)相關(guān)系數(shù)均在0.999 5以上,線(xiàn)性范圍在0~500 mg/L,通過(guò)內(nèi)標(biāo)法測(cè)定靈芝三萜類(lèi)化合物的含量。

表1 流動(dòng)相梯度洗脫方法Table 1 Mobile phase gradient elution program
3 種標(biāo)準(zhǔn)品的HPLC如圖1所示,靈芝酸A的保留時(shí)間為35.1 min,靈芝酸F的保留時(shí)間為53.0 min,靈芝醇B的保留時(shí)間為86.5 min。

圖1 靈芝三萜類(lèi)化合物標(biāo)準(zhǔn)品液相色譜圖Fig. 1 Liquid chromatogram of triterpenoid standards
1.3.3 SMB初始參數(shù)確定
按1.3.1節(jié)B組的方法萃取靈芝,所得萃取液作為SMB的進(jìn)樣原料。選取SMB系統(tǒng)8 只管柱中的1只管柱作為HPLC的分析柱,將C18填料填充于1.0 cm×25 cm的不銹鋼管柱中。采用95%乙醇溶液及0.01%鹽酸溶液作為流動(dòng)相。考察不同流動(dòng)相配比(乙醇溶液與鹽酸溶液體積比為95∶5、90∶10、85∶15、50∶50、45∶55、40∶60)對(duì)分離結(jié)果的影響,從而選擇適合SMB系統(tǒng)的流動(dòng)相。
1.3.4 SMB參數(shù)優(yōu)化設(shè)計(jì)
以1.3.1節(jié)B組方法所得到的萃取液為進(jìn)樣原料,以C18為固定相,選擇1.3.3節(jié)最佳比例的乙醇-鹽酸溶液作為流動(dòng)相。通過(guò)優(yōu)化SMB操作參數(shù):不同的管柱設(shè)計(jì)、流動(dòng)相配比及多通閥的切換時(shí)間等分離純化靈芝三萜類(lèi)化合物。分離實(shí)驗(yàn)共分兩組進(jìn)行:第1組目的是將低極性雜質(zhì)分離,第2組目的是將高極性雜質(zhì)分離。
1.3.5 三角形理論
SMB參數(shù)設(shè)計(jì)主要包括各出入口端流速的設(shè)定與多通閥的切換時(shí)間,本研究SMB參數(shù)的設(shè)定以三角形理論為基礎(chǔ)[30-32],“三角形理論”法是在線(xiàn)性條件或非線(xiàn)性條件下,對(duì)操作SMB快速選擇合適實(shí)驗(yàn)條件的有用工具。它能用于確定最佳條件,并可通過(guò)公式在高產(chǎn)率和短切換時(shí)間上得到一個(gè)折中方案。其主要參數(shù)mj為第j區(qū)段移動(dòng)相的體積流速與固體體積流速的比值,定義如下:

式中:Qj為j區(qū)段流體的體積流速;VC為空管柱體積(本研究所采用的管柱體積為11.78 mL);tsw為所設(shè)定的多通閥切換時(shí)間周期;εt為管柱的孔隙度,即柱橫截面上流動(dòng)相所占的分?jǐn)?shù),為管柱內(nèi)流動(dòng)相體積與柱總體積之比;Vd為死體積,即管柱中未被固定相占據(jù)的空隙體積。
本研究進(jìn)一步假設(shè)εt=0.35,并忽略死角體積Vd=0。
1.4 數(shù)據(jù)統(tǒng)計(jì)及圖表繪制
SMB結(jié)果數(shù)據(jù)統(tǒng)計(jì)方法:SMB系統(tǒng)平衡穩(wěn)定后,收取一個(gè)循環(huán)周期每個(gè)端口的樣品,旋轉(zhuǎn)蒸發(fā)揮干溶劑后,定容,通過(guò)液相色譜定量分析目標(biāo)組分的濃度。
三角形繪制方法:通過(guò)式(2)[33-34]計(jì)算SMB中各組分的亨利常數(shù)K,假設(shè)實(shí)驗(yàn)中共有兩個(gè)組分A和B,其中A組分為弱滯留性成分,B組分為強(qiáng)滯留性成分,所對(duì)應(yīng)的亨利常數(shù)分別為KA和KB,將兩個(gè)K值標(biāo)注于正方形平面的對(duì)角線(xiàn)上,那么平行于縱坐標(biāo)且通過(guò)KA的直線(xiàn)與平行于橫坐標(biāo)且通過(guò)KB的直線(xiàn)將構(gòu)成一個(gè)三角形區(qū)域,該三角形區(qū)域即為理論上可以分離A和B兩組分的操作區(qū)間。依據(jù)式(1)及管柱設(shè)計(jì)形態(tài)分別求得第2段區(qū)段和第3段區(qū)段的m值,即分別為m2與m3值,然后以m2為橫坐標(biāo),m3為縱坐標(biāo)將每組操作條件標(biāo)在平面上。依據(jù)實(shí)驗(yàn)結(jié)果則可推算出實(shí)際可分離A和B兩組分的操作區(qū)間,以下將以虛線(xiàn)三角形表示。

式中:t為SMB單柱實(shí)驗(yàn)中組分的保留時(shí)間;t0為接管柱后系統(tǒng)的死時(shí)間;td為不接管柱時(shí)系統(tǒng)的死時(shí)間;εt為管柱的孔隙度。
2 結(jié)果與分析
2.1 超臨界萃取條件優(yōu)化結(jié)果
如圖2所示,A組僅在萃取的前0.5 h可以得到訊號(hào)較強(qiáng)的萃取物,但峰響應(yīng)低,目標(biāo)物含量低,因此單獨(dú)使用超臨界二氧化碳無(wú)法有效地萃取靈芝子實(shí)體中的三萜類(lèi)化合物。對(duì)比圖2中A、B組可發(fā)現(xiàn),兩者的HPLC圖譜的信號(hào)數(shù)量與峰形極其相似,說(shuō)明此兩種萃取方式所得到的產(chǎn)物基本一致。B組中各組分的響應(yīng)值明顯高于A組,表明添加乙醇作為夾帶劑更能有效地萃取出靈芝三萜類(lèi)化合物。這可能是因?yàn)殪`芝三萜類(lèi)化合物的極性偏高,更易溶于添加乙醇溶液的超臨界二氧化碳中。因此,后續(xù)SMB實(shí)驗(yàn)中的進(jìn)料選擇B組方式進(jìn)行萃取。

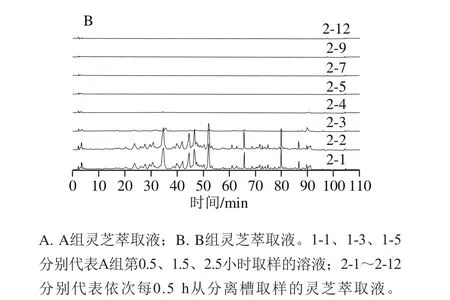
圖2 A組(不添加夾帶劑)和B組(添加乙醇為夾帶劑)萃取液的液相色譜圖Fig. 2 Liquid chromatograms of extracts A (without co-solvent) and B(with co-solvent)
2.2 粗萃物中靈芝三萜類(lèi)化合物含量分析
由圖2B可看出,萃取2 h后大部分物質(zhì)已基本萃取完全。取萃取前3 h中每0.5 h所收集的萃取液進(jìn)行定量濃縮,干燥后可得到每0.5 h粗萃物的量,再根據(jù)目標(biāo)物的濃度可算出每0.5 h粗萃物中目標(biāo)物的質(zhì)量分?jǐn)?shù),如表2所示。計(jì)算結(jié)果表明2-4之后的樣品中靈芝酸A、靈芝酸F及靈芝醇B的質(zhì)量分?jǐn)?shù)均為0或極少,即萃取2 h后目標(biāo)組分已萃取完全,這與圖2B結(jié)果一致。將前2 h收集到的萃取液混合,揮干溶劑共收集得到1.30 g,代表每千克子實(shí)體可以萃取到9.49 g的粗萃物。同時(shí)依據(jù)表2所得質(zhì)量濃度,可計(jì)算得出3 種目標(biāo)成分在粗萃物中的質(zhì)量分?jǐn)?shù):靈芝酸A為4.50%,靈芝酸F為3.39%,靈芝醇B為0.29%。

表2 超臨界二氧化碳混合乙醇萃取靈芝三萜類(lèi)化合物含量Table 2 Contents of triterpenoids extracted by supercritical CO2 extraction using ethanol as co-solvent
2.3 SMB初始參數(shù)確定
單柱實(shí)驗(yàn)中,不同流動(dòng)相配比(95%乙醇溶液與0.01%鹽酸溶液體積比為95∶5、90∶10、85∶15、50∶50、45∶55、40∶60)的分離色譜圖如圖3所示。通過(guò)色譜圖可見(jiàn),乙醇溶液比例高于85%時(shí),可有效地將樣品中的所有物質(zhì)脫附下來(lái),但是當(dāng)乙醇溶液比例低于50%時(shí),便無(wú)法將全部樣品脫附。因此若使用低比例乙醇溶液的流動(dòng)相,在組態(tài)設(shè)計(jì)上需要加入潤(rùn)洗的操作,且潤(rùn)洗操作時(shí)的流動(dòng)相需提高乙醇溶液的比例,才能使固定相成功再生。依據(jù)這些單柱層析圖譜結(jié)果,本研究設(shè)計(jì)出2 組SMB分離實(shí)驗(yàn),通過(guò)去除粗萃液中高極性和低極性雜質(zhì),達(dá)到純化靈芝三萜類(lèi)化合物的目的。其中,第1組SMB的實(shí)驗(yàn)以加入0.01%鹽酸的乙醇溶液作為流動(dòng)相,第2組SMB的分離則以體積比例為40∶60的乙醇溶液與鹽酸(0.01%)混合溶液作為流動(dòng)相。

圖3 不同流動(dòng)相的單柱分離色譜圖Fig. 3 Chromatograms for single column separation with different mobile phases
2.4 第1組SMB分離實(shí)驗(yàn)與結(jié)果
傳統(tǒng)的SMB共有4 個(gè)區(qū)段組成,其中第4區(qū)段主要是利用固體吸附劑將殘留在移動(dòng)相中的弱滯留性物質(zhì)成分清除干凈,避免被移動(dòng)相帶出床體之外繼續(xù)循環(huán)而導(dǎo)致污染。由于本實(shí)驗(yàn)中靈芝粗萃液中的成分種類(lèi)多而復(fù)雜,因此在組態(tài)設(shè)計(jì)上選擇開(kāi)放式系統(tǒng),并且刪除第4區(qū)段,以不回流移動(dòng)相的方式解決弱滯留成分可能循環(huán)累積在系統(tǒng)的問(wèn)題,如圖4所示。設(shè)定SMB組態(tài)為2/3/3,其中共有2 個(gè)入口,即進(jìn)料端(F端)、移動(dòng)相端(D端),以及2 個(gè)出料口,即萃出液端(E端)、萃余液端(R端)。設(shè)定各進(jìn)出口端的流速為D端5 mL/min,F(xiàn)端0.5 mL/min,E端2.083 mL/min,R端3.417 mL/min。

圖4 第1組SMB實(shí)驗(yàn)的管柱組態(tài)設(shè)計(jì)Fig. 4 SMB column configuration design for experimental group 1
當(dāng)系統(tǒng)達(dá)穩(wěn)態(tài)操作后,在2個(gè)出口端收集樣品并分析。在固定流速的情況下,實(shí)驗(yàn)進(jìn)行了不同切換時(shí)間的測(cè)試,由表3可知,在切換時(shí)間為3.4 min時(shí)移除強(qiáng)滯留性的雜質(zhì)效果最佳。且在該條件下,3 種目標(biāo)物的質(zhì)量分?jǐn)?shù)相應(yīng)提高,靈芝酸A提高至5.70%,靈芝酸F提高至4.17%,靈芝醇B提高至0.85%。由于所增加的質(zhì)量分?jǐn)?shù)有限,推測(cè)是因?yàn)榈蜆O性雜質(zhì)的含量不多,所以指標(biāo)成分質(zhì)量分?jǐn)?shù)增加有限。

表3 不同切換時(shí)間的SMB分離結(jié)果(第1組)Table 3 Results of SMB separation at different switching times in experimental group 1
依據(jù)式(1)分別求得m2與m3值,然后以m2為橫坐標(biāo),m3為縱坐標(biāo)繪圖所構(gòu)成的平面圖稱(chēng)其為(m2,m3)平面。如果將SMB實(shí)驗(yàn)所進(jìn)行的操作條件分別標(biāo)示在(m2,m3)平面上,如圖5所示,圖中實(shí)線(xiàn)構(gòu)成的直角三角形代表三角形理論計(jì)算能夠分離低極性雜質(zhì)的操作區(qū)間,其中三萜類(lèi)化合物與雜質(zhì)的等溫吸附常數(shù)分別為0.743與1.107。依據(jù)表3可見(jiàn),在切換時(shí)間周期大約為3.3~3.5 min之間,可以有效移除弱滯留性成分,并依循Yu Hongwei等[33]提出建立最佳操作條件的方法,本研究繪制出圖5虛線(xiàn)所構(gòu)成的三角形。該虛線(xiàn)構(gòu)成的三角形頂點(diǎn)坐標(biāo)為(0.759,1.014),如果在此操作條件下進(jìn)行,假設(shè)切換時(shí)間設(shè)為1.0 min,那么其各出入口端的流量設(shè)定分別為D端12.6 mL/min,F(xiàn)端1.677 mL/min,E端2.389 mL/min,R端11.888 mL/min。若按照最佳條件的流速設(shè)定SMB各進(jìn)出口的流速,以同樣的進(jìn)料質(zhì)量濃度3 937 mg/L進(jìn)行實(shí)驗(yàn),那么可預(yù)測(cè)此SMB設(shè)備平均每天每升填料的處理量,即本SMB系統(tǒng)在移除低極性雜質(zhì)時(shí)固定相的效率為0.061 kg/(L·d)。

圖5 第1組SMB分離實(shí)驗(yàn)三角形理論預(yù)測(cè)Fig. 5 Triangle theory prediction of SMB separation in experimental group 1
2.5 第2組SMB分離實(shí)驗(yàn)與結(jié)果
用1.3.1節(jié)B組方法所得到的萃取液進(jìn)行濃縮,并取濃縮溶液400 mL,再加入600 mL的0.01%鹽酸,過(guò)濾后得第2次進(jìn)料溶液。如圖6所示,本組SMB分離實(shí)驗(yàn)設(shè)定SMB組態(tài)為1-1-3/3,其中共有4 個(gè)入口,即進(jìn)料端(F端)、移動(dòng)相端(D端)、清洗端(Wash端)、潤(rùn)洗端入口(Rinse1端)以及3 個(gè)出料口,即萃余液端(R端),清洗端出口(W端),潤(rùn)洗端出口(Rinse 2端)。移動(dòng)相使用乙醇-鹽酸體積比40∶60,清洗端則使用添加0.01%鹽酸的乙醇溶液,并設(shè)定各進(jìn)出口端的流速為D端3.8 mL/min,F(xiàn)端0.3 mL/min,R端4.1 mL/min,Wash端5 mL/min,Rinse1端5 mL/min。

圖6 第2組SMB實(shí)驗(yàn)的管柱組態(tài)設(shè)計(jì)Fig. 6 SMB column configuration design for experimental group 2
根據(jù)表4可知,在切換時(shí)間為34 min時(shí),R出口端目標(biāo)組分未有響應(yīng),但根據(jù)干質(zhì)量結(jié)果可知R出口端分離出大量物質(zhì),但在本分析方法的檢測(cè)波長(zhǎng)下無(wú)法測(cè)出。同時(shí)根據(jù)Wash端的含量計(jì)算結(jié)果發(fā)現(xiàn)3 種目標(biāo)物質(zhì)量分?jǐn)?shù)大幅度提高,其中靈芝酸A質(zhì)量分?jǐn)?shù)為19.34%,靈芝酸F質(zhì)量分?jǐn)?shù)為15.51%,靈芝醇B質(zhì)量分?jǐn)?shù)為0.74%。這代表大部分的三萜類(lèi)化合物成分是高極性雜質(zhì),少部分為第1組SMB所分離出的低極性雜質(zhì)。因此針對(duì)超臨界流體萃取所得的粗萃物,提高三萜類(lèi)化合物含量的重點(diǎn)在移除高極性的雜質(zhì)。

表4 不同切換時(shí)間的SMB分離結(jié)果(第2組)Table 4 Results of SMB separation at different switching times in experimental group 2
同樣如果將第2組SMB分離實(shí)驗(yàn)所進(jìn)行的操作條件分別標(biāo)示在(m2,m3)平面上,如圖7所示。根據(jù)表4結(jié)果可判定分離的切換時(shí)間周期在34~35 min之間,據(jù)此繪出圖中虛線(xiàn)構(gòu)成的三角形,并計(jì)算出其頂點(diǎn)坐標(biāo)為(6.133,7.662)。如果在此操作條件下進(jìn)行,假設(shè)切換時(shí)間設(shè)定為1.0 min,那么其各出入口端的流量設(shè)定分別為D端64.8 mL/min,F(xiàn)端11.709 mL/min,E端13.712 mL/min,R端62.796 mL/min,并以進(jìn)料濃度3 937 mg/L進(jìn)行實(shí)驗(yàn),預(yù)測(cè)此本SMB系統(tǒng)在移除高極性雜質(zhì)時(shí)固定相的效率為0.423 kg/(L·d)。

圖7 第2組SMB分離實(shí)驗(yàn)三角形理論預(yù)測(cè)Fig. 7 Triangle theory prediction of SMB separation in experimental group 2
3 結(jié) 論
SFE在加入乙醇作為夾帶劑后可有效從靈芝子實(shí)體萃取出靈芝三萜類(lèi)化合物,且萃取物中三萜類(lèi)化合物含量高。SMB的實(shí)驗(yàn)顯示:使用酸性流動(dòng)相可以有效移除非三萜成分;靈芝粗萃液中高極性雜質(zhì)含量較低極性雜質(zhì)含量多。SMB可有效地移除靈芝粗萃液中的雜質(zhì),靈芝三萜類(lèi)化合物從進(jìn)料溶液中質(zhì)量分?jǐn)?shù)為8.51%提高至35.59%,大幅提高了靈芝酸的質(zhì)量分?jǐn)?shù)。輔以三角形理論所建立的最佳操作條件,預(yù)測(cè)了本SMB系統(tǒng)在移除低極性雜質(zhì)時(shí)固定相的效率為0.061 kg/(L·d),而移除高極性雜質(zhì)時(shí)固定相的效率為0.423 kg/(L·d)。
猜你喜歡
作文·小學(xué)低年級(jí)(2025年2期)2025-02-13 00:00:00
小雪花·小學(xué)生快樂(lè)作文(2024年11期)2024-12-31 00:00:00
作文·小學(xué)低年級(jí)(2024年2期)2024-04-29 00:00:00
作文·小學(xué)低年級(jí)(2023年3期)2023-04-29 00:00:00
小獼猴智力畫(huà)刊(2022年9期)2022-11-04 02:31:42
小主人報(bào)(2022年4期)2022-08-09 08:52:06
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
