基于SSR分子標記的藥用黃芪遺傳多樣性與遺傳結構分析
2019-11-07 10:41:36劉亞令耿雅萍解瀟冬張鵬飛
草地學報 2019年5期
關鍵詞:分析
劉亞令,耿雅萍,解瀟冬,王 芳,張鵬飛
(1. 山西農業大學生命科學學院,山西 太谷,030801; 2. 山西農業大學園藝學院,山西 太谷,030801)
藥用黃芪(AstragaliRadix)為豆科植物蒙古黃芪(Astragalusmembranaceus(Fisch.) Bge.var.mongholicus(Beg.) Hsiao)與膜莢黃芪(Astragalusmembranaceus(Fisch.) Bge.)的干燥根[1]。其中野生蒙古黃芪主要分布于山西、內蒙古、甘肅、青海等高海拔、半干旱地區,道地藥材產區以山西渾源、天鎮等北部地區產的蒙古黃芪為優,其次是內蒙古的固陽縣、武川縣等地的蒙古黃芪[2]。野生膜莢黃芪不為主流商品,在中國西南、西北地區以及東北地區的黑龍江、遼寧、吉林地區均有分布[3]。黃芪味甘、性溫,具有補氣、利水、排毒等功效,為常用大宗藥材之一,已擁有2000多年的藥用歷史,是中醫方劑中常用的補氣藥物[4]。目前黃芪不僅用于藥用,還在食療及保健的開發和利用方面也得到了廣泛應用[5]。但隨著黃芪資源市場需求量的增大,造成近幾年野生黃芪的過度采挖,野生黃芪資源幾近枯竭,大面積的成規模的野生黃芪分布已十分少見,大多只是零星分布[2],人工栽培黃芪成為了市場上的主力軍,但人工栽培黃芪種質資源混雜,規范化種植管理和生產水平較低,常會造成其質量及藥用療效的不穩定,因此亟需進行規范化育種以保護黃芪種質資源。研究遺傳多樣性有助于人們更清楚地理解生物多樣性的起源和進化,并為植物分類提供一定的理論依據,進而為植物育種和保護策略的制定奠定基礎[6-7]。
目前,隨著分子標記技術的發展,多種分子標記已被廣泛的用于植物遺傳多樣性分析[8-9]、親緣關系鑒定[10]、核心種質構建[11]、遺傳圖譜的構建[12]等,其中簡單重復序列(Simple Sequence Repeat,SSR)分子標記為共顯性標記,因其具有操作方便、重復性好、多態性高等優點[13],成為理想有效的分子標記之一,現已經成功應用在甘草(GlycyrrhizaL.)[14-15]、黨參(Codonopsis)[16]、黃芩(Scutellariabaicalensis)[17]等多種藥用植物的遺傳多樣性研究及育種中。
因此,本試驗利用SSR分子標記對黃芪主產區山西、內蒙、吉林不同產地的兩種藥用黃芪進行遺傳多樣性及居群遺傳結構分析,了解不同居群不同物種藥用黃芪的遺傳多樣性水平、遺傳分化、變異程度以及居群結構組成,為今后黃芪種質選育利用及保護策略的制定提供一定的參考。
1 材料與方法
1.1 材料的收集
本試驗共采集了380份藥用黃芪材料,其中蒙古黃芪285份,膜莢黃芪85份,涵蓋山西(A)、內蒙古(B)、遼寧(C)、吉林(D)、黑龍江(E)5個省份17個產地共22個自然居群,每個居群進行GPS定位,并對收集到的每個樣本嫩葉保存于裝有硅膠的自封袋中,帶回實驗室進行DNA提取。地理分布圖及采樣詳細信息見圖1及表1。
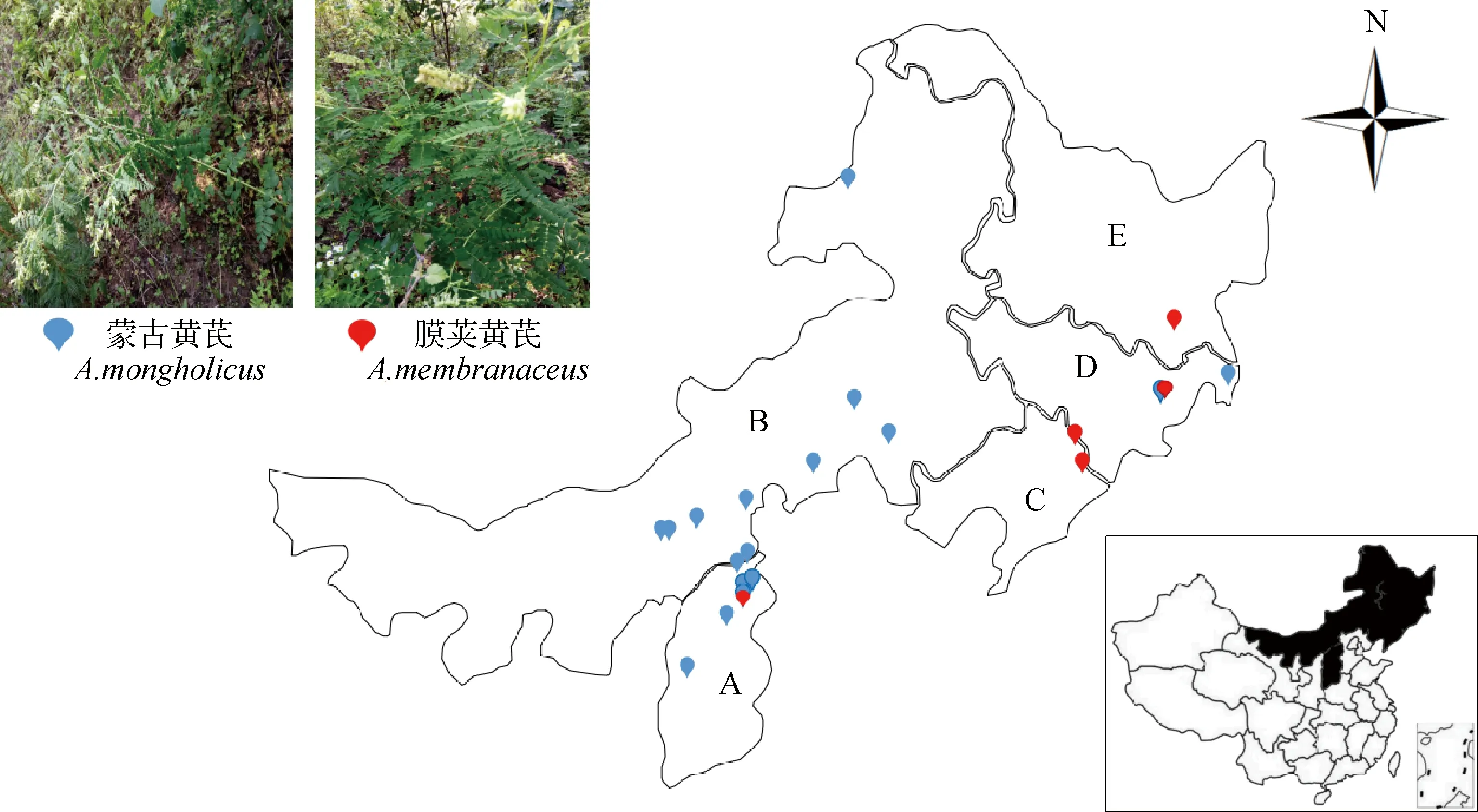
圖1 黃芪采樣地理分布圖
1.2 基因組總DNA提取
采用十六烷基三甲基溴化銨(hexadecyltrimethylammonium bromide,CTAB)法[18]對所有居群中的每個單株樣本材料分別進行基因組總DNA的提取,利用核酸蛋白檢測儀及瓊脂糖凝膠電泳對所提取的DNA濃度及質量進行檢測,將DNA終濃度為調至50 ng·ul-1,并置于—20℃保存以備后用。

表1 黃芪材料來源信息
1.3 PCR擴增及檢測
本實驗根據SSR通用性特征,選用本實驗組所開發的同科植物甘草的64對SSR引物[19],進行多態性的篩選,最終確定10對多態性高、重復性好的SSR引物用于試驗(表2),引物合成由上海生工生物工程有限公司完成。PCR擴增體系參照劉亞令前期研究[20],擴增產物進行Fragment AnalyzerTM全自動毛細管電泳檢測。
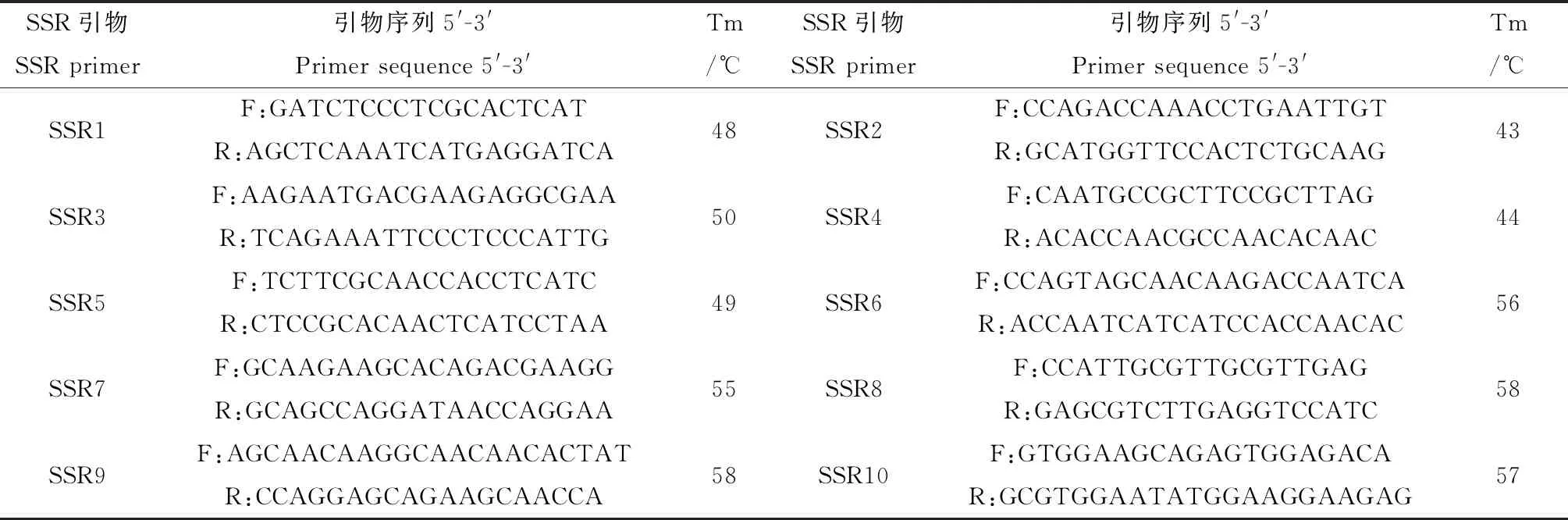
表2 SSR引物序列信息
1.4 SSR數據分析
對毛細管電泳檢測結果進行擴增片段大小的判讀并記錄。利用PowerMarker V3.25軟件[21]計算SSR位點的多態信息含量(PIC),利用GenAlEx 6.503軟件[22]進行遺傳多樣性及遺傳分化分析:主要參數有等位基因數(Na)、有效等位基因數(Ne)、Shannon’s信息指數(I)、Nei’s遺傳多樣性(H)、居群內近交系數(Fis)、總居群近交系數(Fit)、遺傳分化系數(Fst)、基因流(Nm)、期望雜合度(He)、觀測雜合度(Ho);利用GenAlEx 6.503對居群內及居群間分子方差檢驗(AMOVA)及主坐標分析(PCoA);利用MEGA 7.0軟件[23]基于居群間Nei’s遺傳距離[24]構建UPGMA聚類樹;使用Structure V2.3.4[25]軟件分析居群的遺傳結構,其中群體數目(K)設為2~10,對每個K值模擬運算10次,設不作數迭代(length of burn-in period)開始時的馬爾科夫鏈蒙特卡洛(markov chain monte carlo,MCMC)為10 000次,不作數迭代后的MCMC為100 000次,將Structure的運行結果導入在線程序STRUCTURE HARVESTER(http://taylor0.biology.ucla.edu/structureharvest/)進行最佳群體數的預測[26]。
2 結果與分析
2.1 SSR擴增產物的多態性
本試驗利用篩選出的10對SSR引物對380個黃芪樣本的基因組DNA進行SSR-PCR擴增,通過毛細管電泳檢測,擴增結果在150~500 bp之間(圖2)。對黃芪10對SSR位點的遺傳多樣性進行分析,其結果如表3所示:10對引物均具有較高的多態性,其多態信息含量范圍(PIC)為0.698(SSR4)~0.930(SSR6),平均為0.827;等位基因數(Na)為5.455(SSR1)~11.182(SSR6,SSR9),平均為7.818;有效等位基因數(Ne)為3.063~6.527,平均為4.153;Shannon’s信息指數(I)為1.262(SSR2)~2.040(SSR9),平均值為1.573;Nei’s遺傳多樣性指數(H)為0.622(SSR2)~0.820(SSR9),均值為0.706,以上結果均表明篩選的10對SSR引物具有較高的多態信息含量。
2.2 物種遺傳多樣性分析
在物種水平上,分別對蒙古黃芪與膜莢黃芪進行遺傳多樣性分析(表4),結果顯示:蒙古黃芪與膜莢黃芪Shannon’s信息指數(I)分別為I=2.241,I=1.982,平均為2.112;Nei’s遺傳多樣性指數(H)分別為H=0.804,H=0.757,均值為0.781,總體來說兩種黃芪具有較高的遺傳多樣性,其中蒙古黃芪的遺傳多樣性高于膜莢黃芪的遺傳多樣性。此外蒙古黃芪、膜莢黃芪的居群觀測雜合度均小于期望雜合度,且固定指數F均大于零,表明兩種黃芪居群存在一定的雜合子缺陷。
注:等位基因數(Na)、有效等位基因數(Ne)、Shannon’s信息指數(I)、Nei’s遺傳多樣性(H)、期望雜合度(He)、觀測雜合度(Ho)、多態信息含量(PIC)
Note:Na:Average allele number,Ne:Average effective allele number,I:Average shannon′s information index,H:Average Nei′s genetic diversity index,He:Average expected heterozygosity,Ho:Average observed heterozygosity,PIC:Polymorphism information content
注:等位基因數(Na)、有效等位基因數(Ne)、Shannon’s信息指數(I)、Nei’s遺傳多樣性(H)、觀測雜合度(Ho)、期望雜合度(He)、固定指數(F)
Note:Na:Average allele number,Ne:Average effective allele number,I:Average shannon's information index,H:Average Nei's genetic diversity index,Ho:Average observed heterozygosity,He:Average expected heterozygosity,F:Fixation index
2.3 物種遺傳分化分析
分別對兩種黃芪進行遺傳分化分析(表5),結果顯示:蒙古黃芪與膜莢黃芪居群內近交系數(Fis)分別為0.350±0.132,0.387±0.141;總居群近交系數(Fit)分別為0.425±0.122,0.421±0.133;居群間遺傳分化系數(Fst)分別為0.134±0.016,0.058±0.006,表明蒙古黃芪居群間相比膜莢黃芪居群間具有較大程度的遺傳分化,而兩種黃芪種內遺傳分化系數(Fst)分別為0.134±0.016,0.058±0.006,表明蒙古黃芪居群內的遺傳分化(13.4%)高于膜莢黃芪居群內的遺傳分化(5.8%);此外,對兩種黃芪22個居群總的遺傳分化分析得到居群間的遺傳分化系數(Fst)為0.154±0.020,表明黃芪遺傳分化有84.6%來自居群內,有15.4%來自居群間;黃芪居群總的基因流Nm為1.719(Nm>1),表明黃芪居群間基因流處于較高水平。

表5 黃芪種遺傳分化分析
注:居群內近交系數(Fis)、總居群近交系數(Fit)、遺傳分化系數(Fst)、基因流(Nm)
Note:Fis:Inbreeding coefficient within population,Fit:Inbreeding coefficient in total population,Fst:Genetic differentiation coefficient,Nm:Gene flow
2.4 居群聚類分析及Mantel檢測
利用MEGA 7.0軟件基于Nei’s遺傳距離對黃芪22個居群進行了UPGMA聚類分析(圖3),結果顯示:蒙古黃芪與膜莢黃芪首先按物種在0.92處分為兩大類,其中蒙古黃芪這一類中在0.65處又以地域特征分為兩組,第一組中主要是來自內蒙古地區的樣本,第二組則主要是來自山西地區的樣本。

圖3 基于Nei’s遺傳距離的黃芪物種22個居群UPGMA聚類分析
2.3 居群主坐標分析
主坐標分析(Principal Coordinates Analysis,PCoA)是基于居群間遺傳距離得到的一系列的特征值和特征向量進行排序后,繪制的二維或三維坐標圖,從而研究數據相似性或差異性的一種可視化方法。為了進一步了解黃芪不同居群間在遺傳上的差異及相似性,本試驗基于Nei’s遺傳距離對黃芪22個居群進行PCoA主坐標分析,結果得出占總變異貢獻率較多的三個主坐標特征值共為69.59%,其中第一主坐標占總變異的36.5%,第二主坐標解釋了總變異的22%,第三主坐標解釋了總變異的11.09%。二維坐標圖(圖4)結果顯示:黃芪22居群被分為兩大組,其中,膜莢黃芪5個居群分為一組,蒙古黃芪的17個居群分為另一大組,這與UPGMA聚類結果一致。對其前三個主坐標作三維主坐標分析圖(圖5),可以看出其分類結果與二維主坐標分析結果一致,但在二維的基礎上增加了第三主坐標,可以從空間位置上更直觀的了解22個居群間親緣關系的遠近。
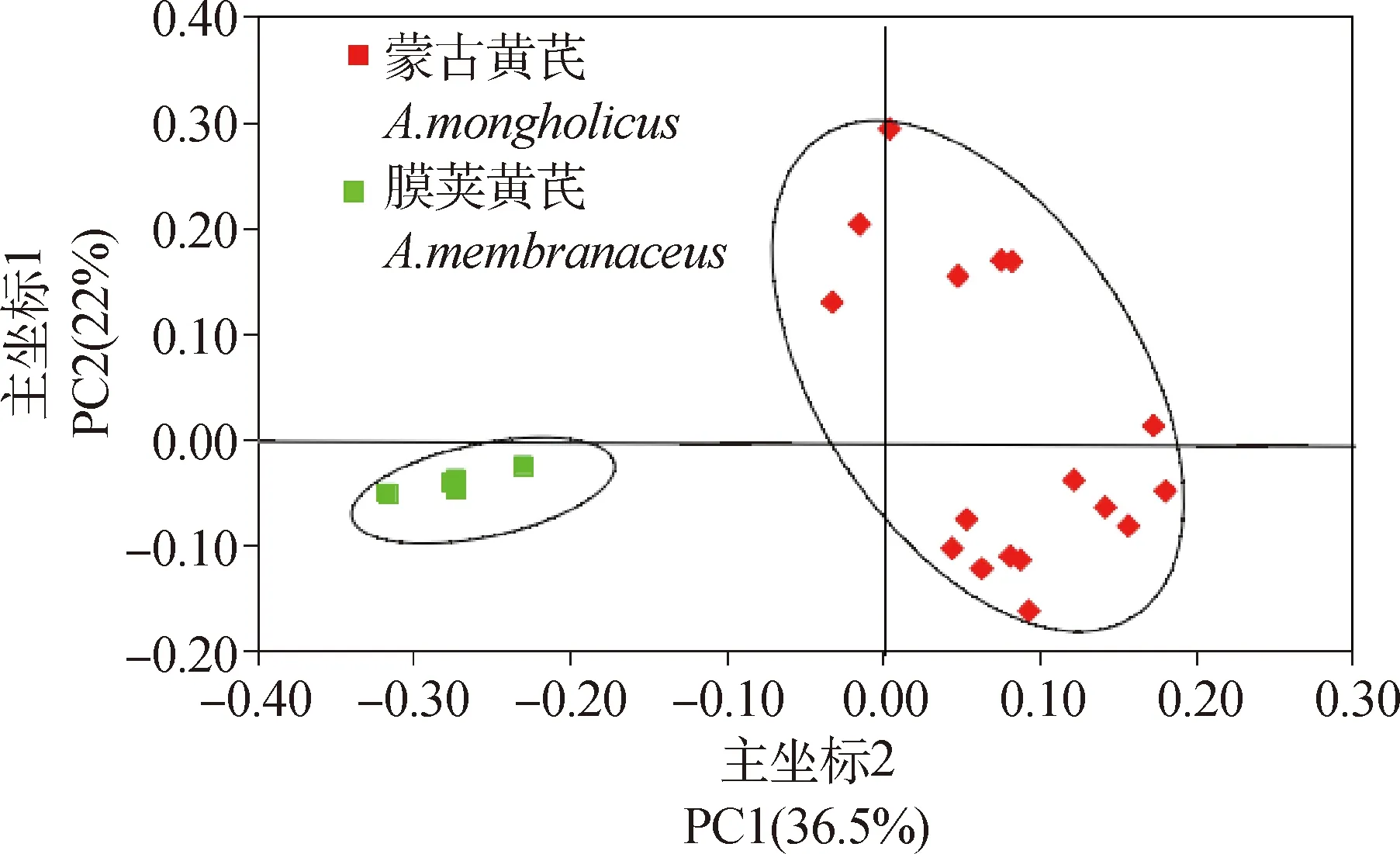
圖4 黃芪22居群主坐標分析的二維散點圖

圖5 黃芪22居群主坐標分析的三維散點圖
2.3 居群遺傳變異分析
遺傳變異水平能夠在一定程度上反映群體在面對不同氣候及生存環境下的適應能力。本試驗利用GenAlEx 6.503軟件對蒙古黃芪及膜莢黃芪居群間、居群內的遺傳變異進行分子方差分析(Analysis of Molecular Variance,AMOVA)(表6),結果得出:黃芪種間遺傳變異為9%,種內居群間的遺傳變異為7%,居群內的遺傳變異為83%,表明遺傳變異主要發生在居群內。這與其遺傳分化分析結果一致(表4),即居群間的遺傳分化(15.4%)小于居群內的遺傳分化(84.6%)。
2.4 居群遺傳結構分析
利用STRUCTURE軟件分析黃芪22個居群遺傳結構(圖6),其中將分組數K設為2-10,每個K值重復10次,并對其作K與Δ K的關系圖(圖6-A),當Δ K達到最大值時(Δ K=737.218),K=3,表明可以將黃芪22個居群分為3個亞群(圖6-B),其中膜莢黃芪為一個亞群(綠色),蒙古黃芪被分為兩個亞群,一亞群主要包括內蒙古地區的的7個居群以及山西地區的3個居群(紅色),第二亞群為山西地區的4個居群(GPJT,GZQL,GXXT,GDXY)及內蒙的兩個居群(GHDM,GMZL)(藍色)。此結果與UPGMA聚類及主坐標分析PCoA結果一致。

表6 黃芪種間及居群分子方差分析(AMOVA)
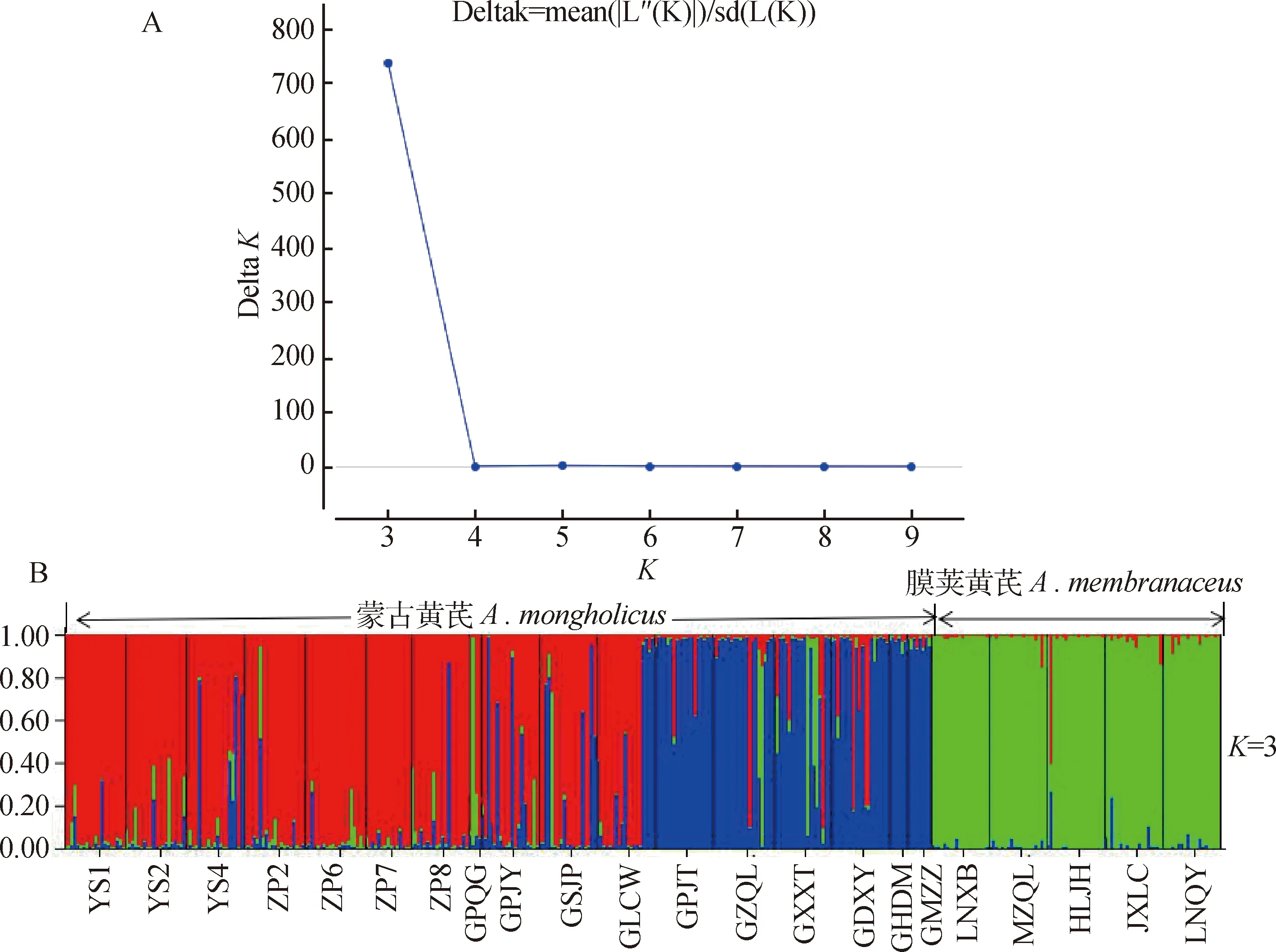
圖6 黃芪22個居群的遺傳結構分析
3 討論
多態信息含量(PIC)是衡量微衛星位點多態性高低的較好指標,一般來說,當PIC>0.5時,具有高度多態性;當0.25 評價藥用黃芪種質資源的遺傳多樣性,對充分利用開發及保護黃芪種質具有重要意義,前人曾利用不同的分子標記進行了不同產地黃芪的遺傳多樣性分析,如張茹[28]利用ISSR對14個產地共183個黃芪樣進行遺傳多樣性分析,得出蒙古黃芪的遺傳多樣性(H=0.336,I=0.497)略高于膜莢黃芪(H=0.311,I=0.466);王傲[29]利用SSR分子標記對162個蒙古黃芪與186個膜莢黃芪的遺傳多樣性結果顯示:蒙古黃芪遺傳多樣性(H=0.822)高于膜莢黃芪(H=0.689);王含彥[30]利用RAPD技術對來自四川、河北、山西、甘肅四個省份的共15份黃芪的遺傳多樣性得出平均Shannon’s信息指數(I)為0.225。本試驗對黃芪主產區山西、內蒙、吉林等地區22個居群共380個樣進行遺傳多樣性分析,結果同樣得出黃芪具有豐富的遺傳多樣性(H=2.112,I=0.781),蒙古黃芪的遺傳多樣性(I=2.241,H=0.804)要高于膜莢黃芪遺傳多樣性(I=1.982,H=0.757),表明了蒙古黃芪作為膜莢黃芪的變種,在不斷地生長進化中,積累了豐富的等位變異基因,比膜莢黃芪有較高的環境適應能力,具有豐富的遺傳育種資源及進化潛力。而本試驗得到的黃芪遺傳多樣性相比前人的研究數值都較高,可能是由于本試驗選用的試驗材料多,產地跨域廣,使其具有高的遺傳多樣性水平。 本試驗基于Nei’s遺傳距離,采用非加權算數平均法(UPGMA法)進行了物種(居群)間聚類分析,結果得出藥材黃芪首先是按物種明顯劃分,此聚類結果與對其表型觀測結果一致,而在同種物種中居群間的聚類較混雜,沒有完全的按地理來源進行聚類,如蒙古黃芪中來自山西的3個居群(GSJP,GLCW,GPJY)與來自內蒙的7個居群聚為一類,另外來自山西的4個居群(GPJT,GZQL,GXXT,GDXY)及內蒙的2個居群(GHDM,GMZL)聚為一類。可能是因為內蒙及山西地區都是藥材黃芪的道地產區,由于人為原因等使兩地區種質間會發生一定的種質交流,且兩地間的地理環境差異較小,在生長過程中可能會發生同等分化及變異。 蒙古黃芪及膜莢黃芪物種間(內)、居群間(內)進行分子方差分析結果顯示,遺傳變異主要發生在居群內(83%)。根據在黃芪傳粉特性的研究中表明,蒙古黃芪和膜莢黃芪均為自交不親和的異花授粉植物[31-32],這一特性使得黃芪在生長繁育的過程中,居使得群內部發生較多的基因交流及變異,因此,這可能是導致黃芪遺傳變異主要發生在居群內的主要原因。 本試驗所選用的10對黃芪同科植物的SSR引物能夠有效的顯示黃芪基因多態性,可用于黃芪遺傳多樣性研究。對蒙古黃芪、膜莢黃芪22個居群共380個樣本進行遺傳多樣性與遺傳結構分析,結果得出黃芪具有較高程度的遺傳多樣性水平,其中蒙古黃芪遺傳多樣性水平要高于膜莢黃芪,遺傳分化程度高于膜莢黃芪。兩種黃芪的遺傳背景相對單一,且遺傳變異主要發生在居群內;UPGMA聚類分析及STRUCTURE群體遺傳分析可將其分為3組,分類結果具有一定的地域性。以上研究結果對藥用黃芪種質資源的有效利用、遺傳多樣性的保護及育種開發優質黃芪種質資源提供一定的理論基礎。4 結論
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
