自由基濃度測定方法的再驗證及其在煤化學中應用
2019-11-22 08:24:24呂海燕方正美張媛媛錢虞峰潘鐵英張德祥
燃料化學學報 2019年11期
呂海燕, 方正美, 張媛媛, 錢虞峰, 潘鐵英, 張德祥,*
(1. 華東理工大學 資源與環境學院, 上海 200237; 2. 華東理工大學 分析測試中心, 上海 200237)
基于中國富煤、貧油、少氣的能源稟賦特點,煤炭作為中國主體能源的地位在很長一段時間內都不會發生根本性轉變,但傳統的直接燃燒煤的利用方式,不僅對煤的利用率低,而且燃燒產生的污染物很多,如SO2、CO、CO2、懸浮顆粒等,對環境造成嚴重污染,實現煤的清潔高效利用是中國能源經濟可持續發展的關鍵[1-3]。煤液化技術是煤清潔高效利用的一種有效途徑,煤加氫液化反應實質是煤熱解生成自由基碎片的穩定、裂解和縮聚反應過程,了解液化中間產物瀝青質自由基的反應機理,有利于提高液化油收率,提高煤加氫液化工藝的經濟性,而了解不同煤階煤的自由基濃度變化規律,有助于豐富煤化學內容。
自1954年Ubersfeld等[4]發現煤中存在穩定的自由基以來,電子順磁共振(EPR)技術普遍應用于煤及其轉化過程中自由基的研究。1967年,Curran等[5]提出了煤液化反應的自由基機理;隨后,Petrakis等[6,7]研究了煤種和液化條件對自由基形成和變化的影響;Malhotra等[8]對八種不同瀝青質組分進行自由基測試。但由于煤樣和液化中間產物中自由基的復雜性,采用EPR傳統方法檢測樣品自由基濃度一直存在難以準確定量的問題。Rudnick等[9]用已知自由基濃度的1,1-二苯基-2-苦基肼基(DPPH)為標準樣品,乙醇為溶劑,通過比較相同測試條件下待測樣品與標準樣品的EPR譜二次積分面積的方法得到煤液化產物的自由基濃度,但乙醇溶劑極性較大,能吸收微波信號,導致結果不準確。Liu等[10,11]直接采用固態DPPH為標準樣品,但DPPH的信號非常強,要求的樣品量非常少,造成的稱量誤差很大。鄭榕萍等[12]建立了一種新的定量測定煤中自由基濃度的方法,該方法將DPPH標準樣品稀釋在硫酸鈣粉末中,制得一系列濃度的標準樣品,以標準樣品的自由基數為橫坐標,EPR譜二次積分面積為縱坐標建立標準曲線測定煤中自由基的濃度,該方法成功解決了溶劑效應、稱量誤差大、單點法準確度低等問題,但由于樣品的EPR譜二次積分面積易受到儀器不穩定性和樣品管差異性等因素的影響,使結果產生一定誤差。
本實驗在鄭榕萍等[12]建立的標準曲線法的基礎上,對其進行優化,以不同質量分數的DPPH標準樣品和基準樣的EPR譜二次積分面積比值作為橫坐標,不同質量分數DPPH標準樣品的自由基數為縱坐標建立標準曲線,其中,基準樣是德國Bruker公司提供的g-marker樣品。以標準樣品和基準樣的EPR譜二次積分面積比值作為自變量,相對于直接以標準樣品的二次積分面積作為自變量,可以減小儀器不穩定性和樣品管差異性對測定結果的影響。并對標準曲線法的準確性、重復性和復現性進行再驗證,然后應用新的標準曲線測試不同煤化程度煤樣和新疆黑山煤直接加氫液化中間產物瀝青質的自由基濃度,以助于更好地了解煤變質過程中煤大分子縮聚反應過程,以及煤加氫液化過程中的自由基反應機理。
1 實驗部分
1.1 樣品的制備
1.1.1 標準樣品的制備
定量稱取DPPH(純度>98%,sigma-aldrich)和無水硫酸鈣,混合研磨30 min,配制成一系列質量分數的標準樣品,分別用棕色瓶密封好存放于冰箱中,2-8 ℃保存備用。
1.1.2 煤樣的制備
本實驗所用的煤樣為云南小龍潭褐煤(XLT)、新疆淖毛湖褐煤(NMH)、新疆黑山長焰煤(HS)、內蒙古補連塔不粘煤(BLT)、江蘇大屯氣煤(DT)、淮北臨渙肥煤(LH)、山西介休焦煤(JX)、河南瘦煤(HN)、山西晉城無煙煤(JC),各煤樣的工業分析和元素分析見表1。原煤先進行破碎研磨至0.178 mm以下,在溫度為80 ℃的真空環境下干燥24 h,密封裝置于硅膠干燥器中備用。

表 1 煤樣的工業分析和元素分析
a: by difference
1.1.3 瀝青質的制備
實驗選用KCF-D型高壓釜作為反應裝置,反應釜體容積為300 mL,實驗初始壓力為6.0 MPa,攪拌速率為500 r/min。稱取20 g HS煤、40 g四氫萘放入反應釜中,并加入催化劑Fe2O3和助劑硫進行反應,到達所需溫度(290-450 ℃)后,恒溫30 min后停止加熱,待溫度降至30 ℃時停止攪拌并收取反應物。先用正己烷對產物進行索氏抽提,得到的不溶物用四氫呋喃進行索氏抽提,再對其可溶物進行旋蒸處理即可得到產物瀝青質,瀝青質產率根據文獻[13]計算得到。
1.2 實驗方法
1.2.1 標準曲線的建立
EPR實驗均用Bruker EMX-8/2.7型電子順磁共振波譜儀,儀器參數為:測試溫度 298 K,微波頻率(9.9±10-8)GHz,微波功率4.05 mW,中心磁場(3520±10-6) G,掃描寬度100 G,時間常數5.12 ms, 掃描時間20.97 s,調制頻率100 kHz,調制幅度1 G。
稱取0.02 g各質量分數的DPPH標準樣品進行EPR測定,記錄其二次積分面積,并將各質量分數的DPPH標準樣品按照下列公式換算為自由基數(N)。
(1)
式中,w為標樣的質量分數,m為稱取的樣品質量,M為DPPH的摩爾質量。
以0.02 g各質量分數的DPPH標準樣品與基準樣的二次積分面積比值作為橫坐標,基準樣的二次積分面積測得為1.0822×108,并以各標準樣品對應的自由基數為縱坐標,建立標準曲線,具體見圖1。由圖1可以看出,標準曲線在測量范圍(3.30×1015-1.51×1017)內線性良好。得到樣品自由基數與EPR譜二次積分面積比值的線性方程如下:
(2)
式中,A為樣品的EPR譜二次積分面積,As為基準樣的二次積分面積,Nc為樣品的自由基數。
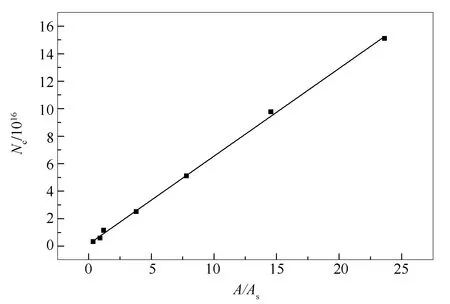
圖 1 DPPH 標準曲線
1.2.2 煤樣和瀝青質中自由基濃度的測定
分別稱取一定質量的HS、NMH、BLT、XLT、DT、LH、JX、HN、JC煤和HS煤不同液化溫度下的瀝青質進行EPR測試,記錄二次積分面積、g因子和峰寬(ΔH),然后根據標準曲線計算所稱取樣品中的自由基數,并按照式(3)計算得到自由基濃度(Ng)。
(3)
2 結果與討論
2.1 標準曲線法的準確性再驗證
稱取一定量的DPPH和無水硫酸鈣,配成質量分數為0.32%的標準樣品,再稱取不同質量的標準樣品進行EPR測試,根據標準曲線得到其自由基數,并與按照式(1)計算得到的理論值進行比較。表2為五份不同質量標準樣品的自由基數實測值和理論值比較結果,結果顯示,每一份標準樣品的自由基數實測值和理論值相對誤差(RE)都在5%以內,說明新建立的標準曲線準確性較好。
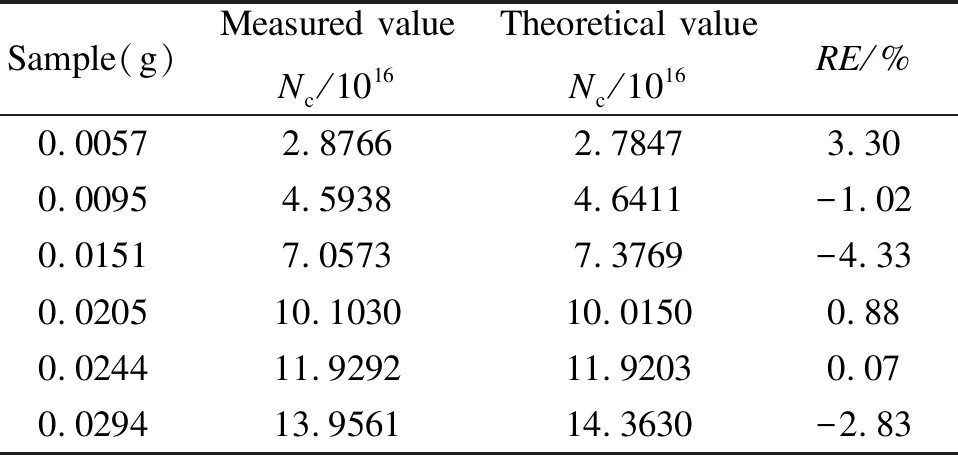
表 2 標準曲線法準確性驗證
2.2 標準曲線法重復性的驗證
各稱取五份相同質量的NMH煤和BLT煤進行EPR測試,根據所得的標準曲線計算得到每份煤樣的自由基數,然后按照式(3)換算為自由基濃度。NMH煤樣稱取的質量為(0.0100±0.0003) g,BLT煤樣稱取的質量為(0.0040±0.0003) g。表3、表4分別為相同質量NMH、BLT煤的EPR測定。
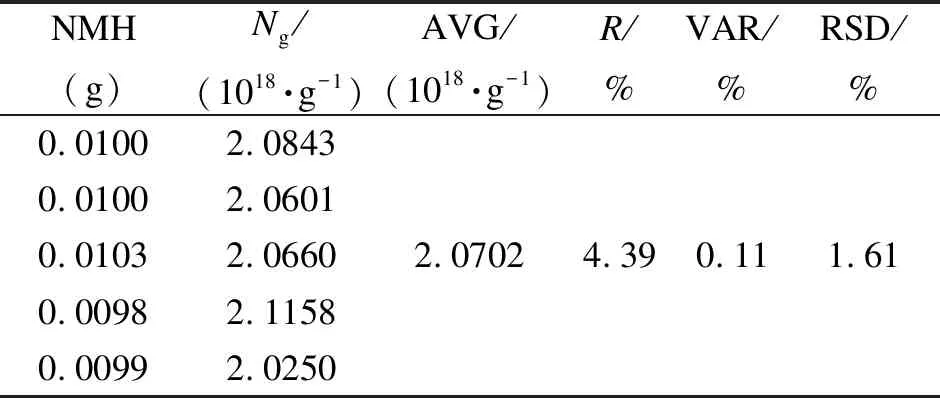
表 3 相同質量NMH煤的EPR測定
表3中五份NMH煤樣的自由基濃度的平均值(AVG)為2.0702×1018/g,相對極差(R)為4.39%,方差(VAR)為0.11%,相對標準偏差(RSD)為1.61%;表4中五份BLT煤的自由基濃度的平均值為1.02827×1019/g,相對極差為3.57%,方差為1.96%,RSD為1.36%。可以看出,新參數的標準曲線法重復性較好。
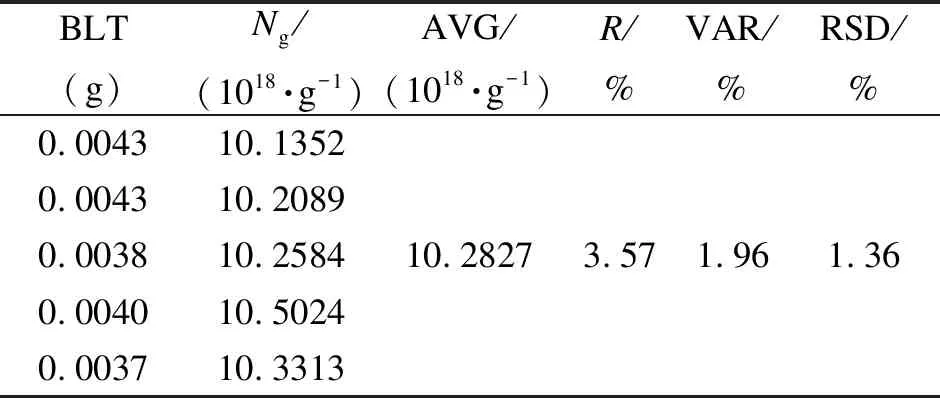
表 4 相同質量BLT煤的EPR測定
2.3 標準曲線復現性的驗證
稱取五份不同質量的NMH煤樣進行EPR測試,并置于棕色瓶中密封存放在冰箱中2-8 ℃保存,相隔一周、兩周、三周、四周之后,再分別進行EPR測試,根據新參數標準曲線計算出自由基數,并換算為自由基濃度。表5是五份不同質量NMH煤在五周內間隔相同的時間測試所得的結果,0.0042 g的NMH煤樣連續五周得到的自由基濃度平均值為2.0938×1018/g,RSD為2.38%;0.0079 g的煤樣五周內測得的五份數據的平均值為2.0389×1018/g,RSD為2.61%;0.0119 g煤樣五份數據的自由基濃度平均值為2.0170×1018/g,RSD為1.76%;0.0165 g煤樣五份數據的自由基濃度平均值為2.0863×1018/g,RSD為2.46%;0.0209 g煤樣的五份自由基濃度平均值為2.0669×1018/g,RSD為1.97%。在標準曲線的測量范圍內,不同質量的NMH煤在五周內間隔相同的時間得到的自由基濃度的RSD都小于3%,表明標準曲線復現性好。從表5還可以看出,五份不同質量NMH煤五周測得的自由基濃度平均值的相對極差為3.73%,方差為0.10%,RSD為1.57%。
進一步證明了用新參數標準曲線法測煤中自由基濃度的準確性和復現性都完全符合測定精度要求。但一定要保證所測樣品的自由基數在標準曲線范圍內,如果超出,其誤差就會很大。如在實驗過程中,稱取0.0158 g的BLT煤樣進行測試時,按照標準曲線計算得到自由基數為2.29604×1017,超出測量范圍,得到的自由基濃度為1.45320×1019/g,與上述重復性試驗得到的BLT煤樣的自由基濃度1.02827×1019/g誤差就比較大。

表 5 標準曲線法復現性測試
2.4 不同煤化程度煤樣的自由基分析
對XLT等九個煤進行EPR測試,獲得自由基濃度、ΔH、g因子三個參數,其結果見表6。
由表6可知,根據新參數標準曲線法計算得到的不同煤化程度煤樣的自由基濃度,從褐煤8.513×1017/g到無煙煤3.37899×1019/g,隨著煤階的升高明顯增大,這與文獻報道相符[14]。在煤化過程中,隨著煤階的增加,由于不斷地脫烷基、脫雜原子等縮聚反應作用,煤中碳含量逐漸增多,H/C逐漸減少,芳環的縮合程度隨之增加,導致小自由基的釋放或偶聯,以及未配對電子在大型結構中的穩定,這是自由基濃度隨著煤階的升高而增大的主要原因,這一趨勢與芳香環共振效應和超共軛效應增加自由基的穩定性認識相一致[15,16]。
峰寬主要表示的是電子從自旋的激發高能級回到低能級的時間即弛豫時間,峰寬和弛豫時間成反比。影響弛豫時間的因素主要有兩點;一是“自旋-晶格”相互作用,即順磁離子和晶格的相互作用;二是“自旋-自旋”相互作用,即未成對電子形成的小磁體之間的相互作用[17]。由表6還可以看出,NMH褐煤的峰寬6.95 Gs最大,JC無煙煤的峰寬3.98 Gs最小,其他煤樣的峰寬在5.39-5.86 Gs呈現分散性。NMH煤樣峰寬最大的主要原因是其未成對電子與氫核的相互作用大大縮短了弛豫時間,導致峰寬變大。JC無煙煤峰寬減小的主要原因是其芳環縮合程度大,未配對電子穩定在縮合苯環的高能級上,導致其弛豫時間延長,峰寬減小[18]。而煤樣的峰寬表現出分散性有兩個主要原因,一是煤中“自旋-晶格”和“自旋-自旋”相互作用的結果;二是線寬與煤中物質的組成有很大的關系,分散性是受煤樣成分混雜的影響[19]。
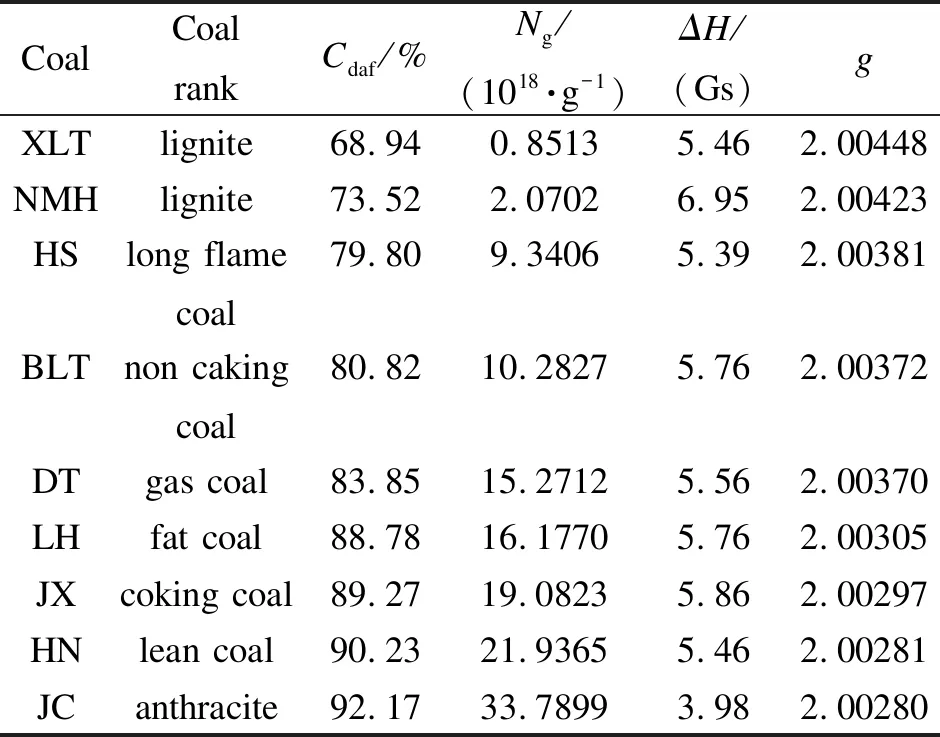
表 6 不同煤化程度煤的EPR測試
g因子能夠反映順磁分子的未成對電子的位置,g值大小主要與自由基種類和雜原子有關,煤中雜原子的存在會使g值增大。據報道,單電子在含氧π軌道上如醚、醌類自由基g值為2.0035-2.0046,含氮自由基g值是2.0031,含硫g值為2.0080-2.0081[20,21]。表6中,隨著煤階的升高,g值逐漸減小,從褐煤2.00448到無煙煤2.00280,主要因素是隨著煤變質程度的增加,碳含量逐漸增加,氧及其他雜原子逐漸減少,如氧含量從XLT褐煤23.27%降至DT氣煤8.96%和JC無煙煤2.79%,且煤內部結構的自由基種類在逐漸減少,所以導致低變質程度煤樣的g值大于中變質程度和高變質程度的煤樣。
2.5 HS不同加氫液化溫度下瀝青質產率和其自由基濃度的關系
圖2是HS煤的瀝青質產率和自由基濃度隨液化溫度的變化。HS煤的自由基濃度為9.3406×1018/g,明顯大于其瀝青質的自由基濃度(7.410×1017/g-1.5793×1018/g),因為原煤經過高溫反應后,側鏈和大分子結構單元之間的橋鍵開始斷裂生成自由基碎片,自由基碎片之間相互反應或與氫結合,部分生成分子量小的液化油,還有一些生成分子量大的瀝青質,但瀝青質的分子量是小于原煤的分子量的,所以自由基濃度也相對應的比原煤小[22]。在液化溫度290-450 ℃,隨著溫度的升高,瀝青質產率逐漸降低,其自由基濃度也呈現下降趨勢。其中,在290-380和410-450 ℃,瀝青質自由基濃度下降的幅度比380-410 ℃下降的幅度小,與瀝青質產率下降的幅度也一致。在290-380 ℃,隨著溫度的升高,加氫反應劇烈,芳環側鏈開始斷裂,生成液化油和氣體等小分子物質,瀝青質產率降低,能在瀝青質大分子共軛結構中離域而穩定存在的自由電子也相對應減少。當溫度達到380 ℃,主要發生自由基碎片的縮聚反應,更多的自由基被穩定下來,生成大分子芳環物質,且溫度越高,初始產生的自由基越多,縮聚反應也越劇烈,自由基濃度下降的更明顯。在410-450 ℃,其自由基產生速率和縮聚反應速率逐漸達到一定的平衡,從而使下降幅度趨于平緩。
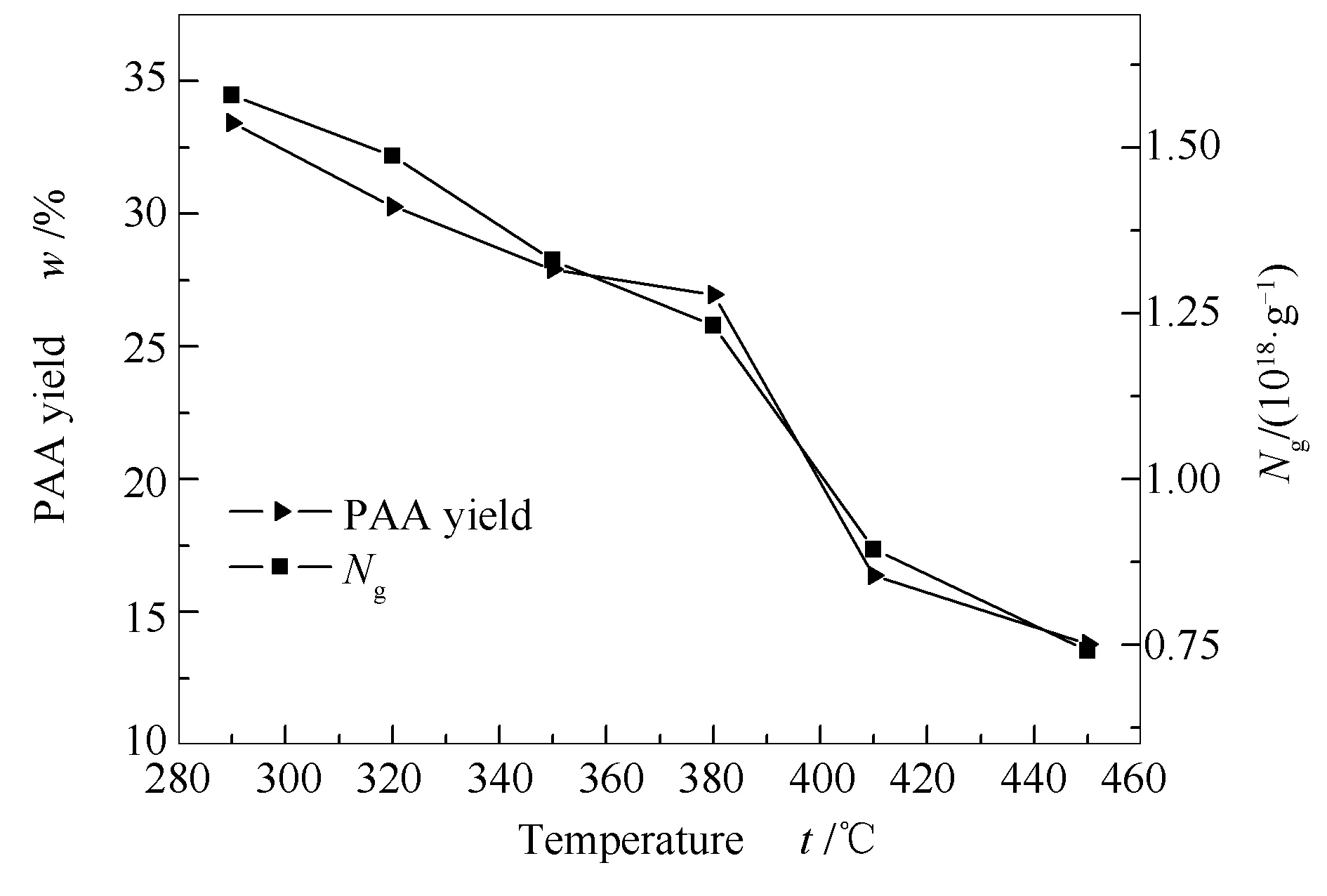
圖 2 HS瀝青質產率和自由基 濃度隨液化溫度的變化
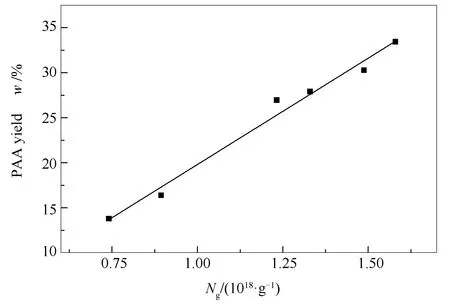
圖 3 HS不同加氫液化溫度的瀝青 質產率和其自由基濃度關系
隨著液化溫度的升高,HS煤瀝青質產率逐漸下降,其自由基濃度也在相應的降低,且兩者降幅一致,可以看出兩者存在一定的相關性。通過曲線擬合得到圖3。瀝青質自由基濃度與產率之間的關系如式(4)所示:
y=23.6349x-3.8167
(4)
式中,y為不同溫度下HS煤加氫液化瀝青質產率,w%;x為對應的瀝青質的自由基濃度,Ng/(1018·g-1);曲線的相關性R2=0.98079。
3 結 論
本實驗以各質量分數DPPH標準樣品和基準樣的二次積分面積比值為新參數建立標準曲線,標準曲線測得樣品的自由基數與理論值相對誤差都在5%以內,重復性和復現性實驗的相對標準偏差都小于3%,三者均滿足分析精度的要求,該方法可靠。
根據新參數標準曲線法測試得到的不同煤化程度煤樣的自由基濃度從褐煤8.513×1017/g逐漸上升到無煙煤3.37899×1019/g,而g值從褐煤2.00448逐漸下降到無煙煤2.00280,這與隨著煤變質程度的加深,雜原子脫除,脂肪碳側鏈斷裂,芳環縮合程度加深有關,豐富了煤化學內容。
將新參數標準曲線法運用到HS煤不同加氫液化溫度下瀝青質自由基濃度的測試,發現在290-450 ℃,瀝青質自由基濃度從1.5793×1018/g下降到7.410×1017/g,與其產率變化趨勢相一致,兩者具有線性相關性,為研究煤加氫液化機理提供技術支撐。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
產品可靠性報告(2017年7期)2017-09-05 09:49:12
專用汽車(2016年4期)2016-03-01 04:13:43
汽車觀察(2016年3期)2016-02-28 13:16:26
