乙肝病毒(HBV)感染對肝細胞癌(HCC)患者A-to-I RNA編輯的影響
2019-12-05 07:23:38馬婷黃海郝沛
復旦學報(醫學版) 2019年6期
馬 婷 黃 海 郝 沛
(1上海大學生命科學學院發育神經生物學實驗室 上海 200444; 2中國科學院上海巴斯德研究所病原大數據實驗室 上海 200031)
肝細胞癌(hepatocellular carcinoma,HCC)是原發性肝癌中最主要的組織學亞型[1]。纖維板層肝細胞癌(fibrolamellar-HCC,FL-HCC)是HCC的一種特殊類型,常見于青年群體,通常不伴隨肝硬化和乙型肝炎病毒(hepatitis B virus,HBV)感染[2-3]。已知HBV感染后會增加HCC的發病率[4]。
RNA編輯是指通過轉錄后修飾引起轉錄RNA序列改變,致使RNA攜帶信息改變的生物學現象[5]。脊椎動物中最常見的RNA編輯類型是腺嘌呤(adenine,A)轉變為次黃嘌呤(inosine,I),即A-to-I RNA編輯。A-to-I RNA編輯發生在具有雙鏈特征的RNA區域。底物RNA在腺苷酸脫氨酶(adenosine deaminases acting on RNA,ADAR)催化下將A水解脫氨基轉化為I,后續翻譯過程中被識別為鳥嘌呤(guanine,G)[6]。在病毒感染期間,不同的病毒與宿主組合后,ADAR可能發揮促病毒或抗病毒作用[7-8]。目前對HCC患者RNA編輯的研究主要集中在癌組織和正常組織間的編輯活性變化[9-10],而HBV感染對HCC患者A-to-I RNA編輯事件的影響尚無研究報道。
我們從數據庫下載了HBV陰性和陽性患者的轉錄組數據[11-12],采用SPRINT軟件[13]鑒定A-to-I RNA編輯事件,比較正常組織和癌組織中HBV陰性、HBV陽性樣本組的ADAR1表達值及共有RNA編輯位點(RNA editing site,RES)的編輯水平差異情況。本研究首次分析了HBV感染的HCC患者A-to-I RNA編輯的變化情況,對深入研究HBV感染對HCC發生發展的分子作用機制有一定的意義。
材料和方法
材料轉錄組數據來自美國國家生物技術信息中心(National Center of B-iotechnology Information,NCBI)的GEO數據庫,數據集編碼為GSE63018和GSE77509。為區分兩套數據,分別用FL-HCC和HCC來表示。FL-HCC數據集為2*50的雙端非特異性測序數據,測序數據來自FL-HCC患者,由于該類HCC的特殊性,8位患者均為HBV陰性。HCC數據集則為2*100的雙端非特異性測序數據,包含19位HBV陽性患者和1位HBV陰性患者。每位患者均有配對的正常和癌癥肝組織的測序數據。
A-to-I RESs的鑒定和注釋采用SPRINT 0.7.16軟件鑒定RNA編輯事件。將匹配到參考基因組的讀段(reads)和未匹配、處理后二次匹配的讀段分為兩條檢測途徑進行位點尋找,因此該法能找出數目更多的位點用于后續分析。在SPRINT中使用Burrows-Wheeler algorithm (BWA)算法[14]進行比對。人類參考基因組和基因組注釋信息版本為hg19。采用Annovar軟件對得到的位點進行注釋[15]。
基因差異分析使用Trim_galore v0.4.3(http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)去除測序質量低于20的讀段,得到清潔數據。采用“-fastqc”參數對清潔數據進行測序質量評估。采用Hisat2 v2.0.5[16]比對到人類參考基因組得到SAM文件。用Samtools v1.5[17]的“samtools view”和“samtools sort”命令將SAM文件轉化為BAM文件并進行排序。使用Picard v1.127軟件(https://broadinstitute.github.io/picard/)去除樣本制備過程中產生的PCR重復序列。HTSeq-0.6.1[18]用于計算基因比對到每個基因的讀段數。使用轉錄組表達定量值CPM(counts per million)來實現基因表達值的標準化,即原始讀段數÷總讀段數*1 000 000。
標準化編輯水平計算編輯水平定義為每個發生A-to-I RNA編輯的讀段數與比對到該位點的所有讀段數的比值。為更好比較不同患者的編輯水平變化,我們選出正常組織和癌組織共有的RES。將共有RES最高的值作為標準,其他RES與之相除,得到標準化編輯水平[19]。
GO富集分析使用R包clusterProfiler軟件[20]對每個患者正常組織和癌癥肝組織的RES所在基因分別進行GO富集分析。找出在HBV陰性和HBV陽性樣本組中均出現的GO富集通路,其中校正后P<0.05的通路為顯著富集通路。
圖形展示和統計學方法使用R開源包ggplot2和GraphPad prism8.0.1繪制圖片。本研究在R環境下進行統計學分析,相關性檢驗使用Kruskal-Wallis檢驗,P值閾值設置為0.1。
結 果
兩套數據集A-to-I RESs概況我們檢測了FL-HCC和HCC數據集中56個樣本的RNA編輯事件,分別找出299 528和122 265 509個RES(表1)。為驗證RES的可靠性,將每個樣本的檢測位點與目前主要的RES數據庫DARNED(http://darned.ucc.ie/)、RADAR(http://rnaedit.com/)、REDIportal(http://srv00.recas.ba.infn.it/py_script/REDIdb/)以及3個數據庫的并集進行比較(表1)。結果發現,FL-HCC數據集與數據庫的平均交集為76.04%,而HCC數據集與數據庫的平均交集僅為52.47%。這是由于數據庫中的位點主要是匹配到參考基因組的讀段所找出的RES,而SPRINT還包含未匹配到基因組而進行二次匹配的讀段所找出的RESs。所以兩套數據集與數據庫的交集并不高,這說明相對于以往的RES鑒定方法[21-22],本研究可檢測出更多的RES來進行后續分析。
為進一步驗證檢測位點的準確性,我們分析了兩套數據集RES的位點分類特征(圖1)。將RES分為Alu區、重復非Alu (repetitive non Alu,REP-NON-Alu)區和非重復(non repetitive,NON-REP)區,FL-HCC和HCC數據集分別有超過72%和82%的位點位于Alu區,兩套數據集RES的分布特征具有一致性。這說明由于Alu區的序列具有相似特征,其在基因和基因間易形成雙鏈RNA,是ADAR酶的理想底物。因此,大部分位點發生在基因組的Alu區域,該現象與已有研究結果一致[23]。大部分位點分布在內含子和基因間區域,其次是3’-UTR和非編碼RNA(non coding RNA,ncRNA)區域,外顯子和5’-UTR區域分布最少。這些分布特性都表明我們所鑒定出的位點具有可信度。
FL-HCC:RNA-seq data collected from GSE63018;HCC:RNA-seq data collected from GSE77509.SPRINT:Number of RESs identified by SPRINT.
ADAR酶表達水平的變化HCC組織中ADAR1表達量升高與RNA編輯水平上調有明顯相關性[9],但HBV感染后HCC樣本ADAR1的表達變化與RNA編輯水平之間的關系尚不清楚。我們將兩套數據集的樣本按照不同組織來源和HBV感染情況,分為HBV陰性正常組織(HBV-N)、HBV陰性癌組織(HBV-T)、HBV陽性正常組織(HBV+N)和HBV陽性癌組織(HBV+T)等4個樣本群體。考慮到兩套數據集的批次效應,我們使用標準化的基因表達值來觀察在不同組織和HBV感染情況下ADAR1的表達變化。在比較HBV-N和HBV+N、HBV-T和HBV+T群體后,我們發現在正常和癌組織中HBV感染均引起ADAR1的表達上調。在HBV陰性群體中,ADAR1在正常組織和癌組織之間無明顯差異。在HBV陽性群體中,RNA編輯酶的整體活性更高,且癌組織的ADAR1表達水平相對于正常組織有明顯上升(圖2A)。這都說明HBV感染促進ADAR1表達,該現象在癌組織中更顯著。此外,我們還觀察到不同樣本群體中ADAR2和ADAR1的表達趨勢并不一致(圖2B)。在HBV陰性群體中ADAR2在癌組織中的表達水平相對于正常組織更高,而HBV陽性群體中則相反。在不同組織中,HBV感染后ADAR2的表達水平更低。總體上,ADAR2的表達值明顯低于ADAR1的表達值,這表明在催化編輯反應中ADAR1發揮主要作用,ADAR2的作用有待進一步分析研究。
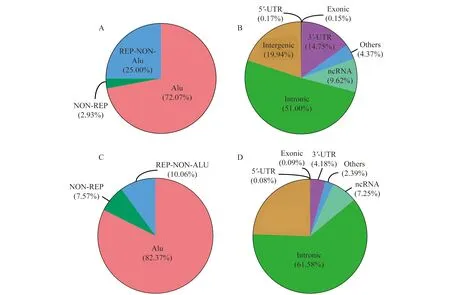
Distribution (A) and genomic distribution (B) of RESs in FL-HCC data set;distribution (C) and genomic distribution (D) of RESs in HCC data set.
圖1 FL-HCC和HCC數據集中A-to-I RESs分布情況
Fig 1 The distribution of A-to-I RESs in FL-HCC and HCC data sets
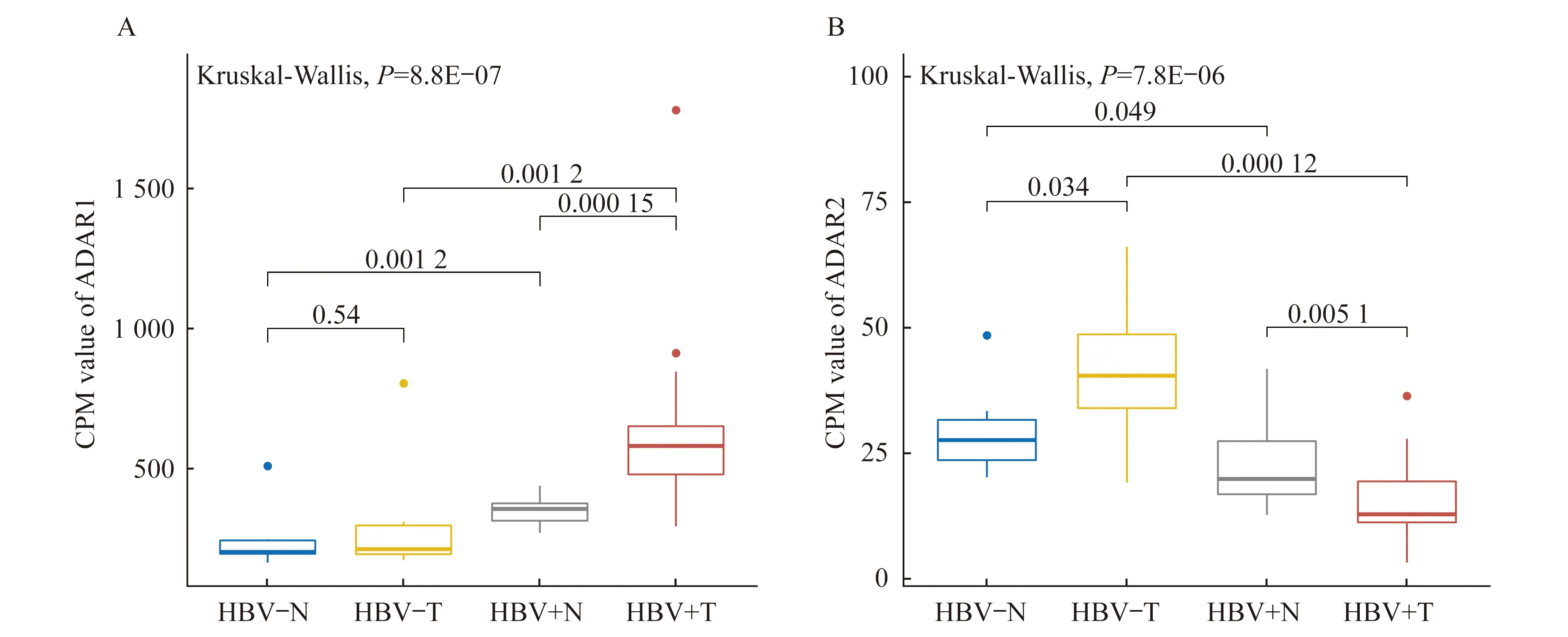
HBV-N:HBV negative and normal tissue;HBV-T:HBV negative and tumor tissue;HBV+N:HBV positive and normal tissue;HBV+T:HBV positive and tumor tissue.CPM:Count-per-million.
圖2 不同HBV感染狀態下正常組織和癌組織中ADAR1(A)和ADAR2(B)的表達水平
Fig 2 The expression levels of ADAR1 (A) and ADAR2 (B) in normal and tumor tissues in response to HBV infection
A-to-I RESs編輯水平的變化由于ADAR1在催化A-to-I RNA編輯反應中起主要作用,進一步分析ADAR1的上調是否導致編輯水平的顯著變化。我們取出每位患者正常組織和癌組織共有的A-to-I RESs后,計算出這些位點的標準化編輯水平。根據共有RESs的編輯水平計算出均值,比較不同感染狀況和不同組織來源樣本群體的RESs編輯水平變化(圖3)。不同感染狀態下樣本的編輯水平在0.30~0.45,多集中在低于0.5的區域中。雖然HBV感染時ADAR1在兩類組織中均顯著上升,但僅在癌組織中發現編輯水平上升(P<0.1)。在HBV陽性群體中,癌組織的編輯水平相對于正常組織有所提升(P<0.1),但在HBV陰性群體中則沒有類似現象。這說明ADAR1表達變化對癌癥樣本和HBV感染樣本的RESs作用更為明顯。
A-to-I RESs所在基因的GO富集情況分析RESs所在基因的GO富集情況,找出在HBV陰性和陽性樣本中均出現的GO富集通路(表2):HBV陰性樣本的編輯基因顯著富集在細胞氨基酸代謝過程通路(P<0.05);HBV陽性樣本的編輯基因則富集在蛋白質絲氨酸/蘇氨酸激酶活性、轉錄后調控基因表達及去磷酸化相關信號通路(P<0.05)。這些通路與細胞增殖、基因調控過程相關。這說明HBV感染使細胞增殖并調控基因表達相關的編輯基因活性發生改變,這可能對HCC的發生造成影響。

HBV-N:HBV negative and normal tissue;HBV-T:HBV negative and tumor tissue;HBV+N:HBV positive and normal tissue;HBV+T:HBV positive and tumor tissue.
圖3 在不同HBV感染狀態下正常和癌癥組織中RESs的編輯水平變化
Fig 3 The editing level of RESs in normal and tumor tissuesin response to HBV infection
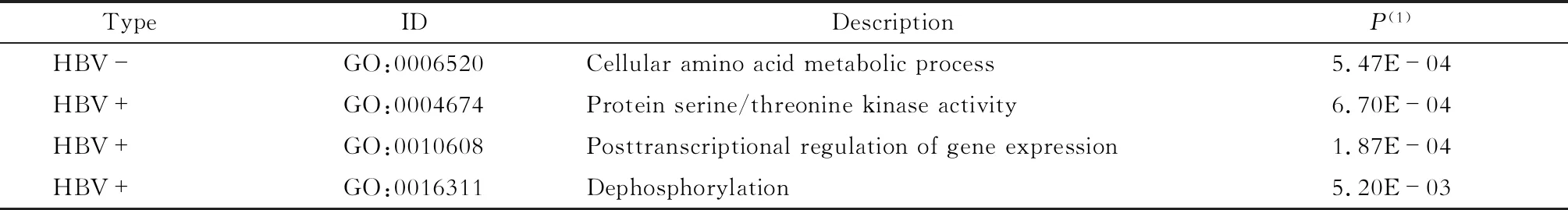
表2 不同HBV感染狀態下RESs所在基因的GO富集通路Tab 2 The GO enrichment pathway of genes with RESs in response to HBV infection
HBV-:HBV negative;HBV+:HBV positive.(1)AjustedP.
討 論
HCC是一種異質性腫瘤,在遺傳信息和表觀遺傳層面表現出復雜多樣的變化。在癌組織中,異常的轉錄后修飾(如RNA編輯),可能導致腫瘤轉錄組的多樣性[23]。RNA編輯在病毒感染過程中同樣起著重要作用[7]。目前對HCC患者RNA編輯的研究多為癌組織相對于正常組織的RNA編輯事件變化,對HBV感染后患者RNA編輯的變化研究較少。
我們使用SPRINT軟件來檢測編輯位點,相對于以往的位點鑒定方法[21],該法能發現更多RES,有利于后續位點的統計分析。通過分析每位患者RES的數目和分布特征,發現兩套數據集的RES數目有明顯差異,數據測序深度大的樣本能檢測到更多的RES,這與數據本身測序深度和讀數長度有關,因而從位點數量分析HBV感染對患者RNA編輯的影響并不合適。通過分析位點在基因組上的分布,發現不同數據集的樣本分布趨勢具有較強的一致性,RES大部分落在內含子區域和基因間區域。這與以往研究結果一致[9]。不同HBV感染狀態下,正常組織和癌組織中ADAR1表達水平均顯著上升。HBV感染后,癌組織中ADAR1表達上調,同時其共同RES的編輯水平也會隨之升高,而在未感染HBV群體中則沒有類似發現。這說明HBV感染極可能通過上調ADAR1表達,對編輯基因的表達活性產生影響。HBV感染樣本的編輯基因顯著富集在基因調控和細胞增殖相關通路,說明編輯基因的活性改變可能影響了細胞的正常生長,進而影響HCC的發生。這提示在治療HBV感染患者的過程中抑制ADAR1的表達對預防HCC的發生可能會起到作用。
本研究收集的數據來自FL-HCC和HCC兩套公開數據集。考慮到不同數據集對后續分析的影響,分別對兩套數據集在基因組上的分布進行了分析,發現它們在基因功能區的分布是一致的。這在一定程度上表明樣本組織來源的不同對RESs分布影響不大。考慮到批次影響,我們在分析基因表達水平時采用標準化基因表達值CPM來衡量ADAR表達水平。在分析RESs的編輯水平時,將正常組織作為背景,選取腫瘤和正常組織共有的RESs來分析,這在一定程度上能夠去除由于樣本的組織類型差異所造成的編輯水平變化。由于患者年齡、性別和病毒感染等因素也可能對分析造成一定的影響,因此我們在選取數據時盡量選擇范圍一致的樣本。由于公共數據庫的數據有限且未感染HBV的臨床樣本不易獲取,本研究收集到的不同HBV感染情況的患者樣本數目并不一致。更多未感染HBV的HCC患者樣本有助于校正分析過程中的偏差。
總之,本研究利用轉錄組數據描述HBV陽性和陰性HCC樣本A-to-I RNA編輯事件的區別,發現HBV感染上調ADAR1的表達,從而改變宿主編輯事件的活性,這對HCC的發生發展可能有促進作用,也為進一步探索宿主RNA編輯對HBV感染的響應機制提供了線索。
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年12期)2019-05-21 02:55:32
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
學苑創造·A版(2015年11期)2016-01-14 09:03:27
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
中國火炬(2010年8期)2010-07-25 11:34:30
