Becker型肌營養(yǎng)不良合并嚴(yán)重心肌損害1例
2019-12-19 04:00:54王舒?zhèn)R牟立欣李光源王永權(quán)馬春燕
中國醫(yī)學(xué)影像技術(shù) 2019年12期
王舒?zhèn)R,牟立欣,李光源,王永權(quán),馬春燕,楊 軍
(中國醫(yī)科大學(xué)附屬第一醫(yī)院心血管超聲科,遼寧 沈陽 110001)
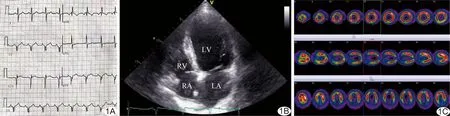
圖1 BMD合并嚴(yán)重心肌損害表現(xiàn) A.心電圖; B.超聲心動圖 (LA:左心房;LV:左心室;RA:右心房;RV:右心室); C.心臟ECT
患者男,17歲,因“四肢無力12年,勞累后氣短1年”于我院就診。患者近12年來無誘因四肢無力,無肌肉痛,無感覺麻木及肌肉萎縮;1年前出現(xiàn)跑步、爬樓梯等運動后氣短,于外院診斷為“擴張型心肌病”,未接受系統(tǒng)治療。查體:神清語明,頸軟,顱神經(jīng)正常,四肢肌力Ⅳ級,球海綿體肌反射(++),Babinski征(-),未見明顯痛覺異常,翼狀肩胛,雙側(cè)弓形足,雙小腿腓腸肌假性肥大,Gowers征(+),輕微步態(tài)搖擺。實驗室檢查:血清天門冬氨酸氨基轉(zhuǎn)移酶64 U/L(15~40 U/L),肌酸激酶(creatine kinase, CK)4 905 U/L(39~308 U/L),乳酸脫氫酶331 U/L(135~225 U/L),CK-MB 41 ng/ml(0~7.2 ng/ml)。心電圖示下壁、側(cè)壁導(dǎo)聯(lián)異常Q波(圖1A)。超聲心動圖:左心室擴大,下壁及側(cè)壁運動減弱,左心室射血分?jǐn)?shù)(31%)減低(圖1B),診斷為擴張型心肌病改變。ECT:左心室前壁、下側(cè)壁、側(cè)壁心肌血流灌注量輕-中度降低,約占左心室面積的15%,左心室下壁及下側(cè)壁室壁變薄,左心室整體收縮功能降低(圖1C);診斷為左心室心肌病變,心肌缺血。肌電圖:右腓腸肌、右股四頭肌呈可疑肌源性損害。基因檢測:假肥大性肌營養(yǎng)不良(Duchenne muscular dystrophy, DMD),Xp21基因3~7號外顯子缺失,其余外顯子未見異常。結(jié)合病史、影像學(xué)表現(xiàn)及基因檢測,綜合診斷為:Becker型肌營養(yǎng)不良(Becker muscular dystrophy, BMD)合并嚴(yán)重心肌損害。
討論進行性肌營養(yǎng)不良(progressive muscular dystrophy, PMD)分類眾多,BMD為最常見的類型之一。BMD為Xp21基因突變所致的X連鎖隱性遺傳病,主要表現(xiàn)為四肢近端進行性肌無力,腓腸肌假性肥大,Gower’s征陽性等。BMD在人群中的患病率約為2.4/100 000,患者癥狀通常較輕,可存活至50~60歲,心肌受累是其最主要的死因。60%~75%的BMD患者可合并心肌受累,發(fā)生心肌受累時年齡多為30歲以上。由于BMD患者骨骼肌受累癥狀較輕,臨床上易忽視,將心肌受累誤診為原發(fā)性心肌病變。本例發(fā)病年齡僅為17歲,四肢無力癥狀較輕,且伴有嚴(yán)重的擴張型心肌病表現(xiàn),在低于20歲的BMD患者中極為少見,因而易誤診,但結(jié)合病史、查體及影像學(xué)檢查,有助于明確診斷。確診BMD的金標(biāo)準(zhǔn)為基因檢測和肌肉活檢。
