基于共價有機骨架-MSPE-HPLC-UV法測定多環芳烴
2019-12-19 07:48:52胡碧清李倩蓮嚴志明
食品與機械 2019年11期
關鍵詞:分析
胡碧清 - 葉 佳 李倩蓮 - 龐 杰 嚴志明 -
(福建農林大學食品科學學院,福建 福州 350000)
多環芳烴(polycyclic aromatic hydrocarbons,PAHs)是重要的環境和食品污染物,通常來自有機物質的不完全燃燒,如煤、汽油、木材、肉類或其他有機食品[1]。其具有潛在的生物蓄積性,致突變、致癌和致畸性,對人類健康和環境構成潛在威脅[2-3]。廣泛分布于環境和食品中的PAHs具有痕量水平的低濃度和低溶解性。因此,建立一種高效、靈敏的PAHs分析方法尤為重要。
目前,樣品前處理技術主要包括固相萃取[4-5]、攪拌棒吸附萃取[6-7]、固相微萃取[8-9]、磁性固相萃取[10-12]等。磁性固相萃取是一種基于固相萃取的分散固相萃取技術,相比于其他前處理技術,只需使用少量的吸附劑和較短的平衡時間就能夠實現低濃度的微量萃取,具有非常高的萃取能力和萃取效率。磁性固相萃取的核心是磁性納米粒子,具有大的比表面積和獨特的物理化學性質,特有的超順磁性有助于通過磁鐵方便地分離和收集。近年來,基于磁性納米粒子的技術在分離領域中備受關注[13-15]。石墨烯、碳納米管、金屬—有機框架材料、分子印跡聚合物、共價有機骨架等材料用于制備磁性復合材料并應用于磁性固相萃取。
共價有機骨架材料(covalent organic framework,COFs)是一類完全由C、H、O、N等輕元素組成并且通過強共價鍵連接的有機晶體材料。COFs作為晶型多孔材料,具有低密度、高比表面積、良好的化學及熱穩定性等特點而備受關注和研究[16-17]。在過去的10余年中,COFs在各個領域得到了廣泛的應用,如氣體存儲與分離[18-20]、催化[21-23]、傳感[24-25]、吸附劑[26-28]。近年來,COFs在樣品前處理方面展現了巨大的應用潛力[29-30],尤其是磁性COFs材料可以克服因COFs材料密度輕及粒徑小所引起的使用不便的問題。目前,磁性COFs的制備及在磁性固相萃取應用仍存在發展階段,磁性COFs的制備存在操作繁瑣、過程冗長、制備條件苛刻等問題。
試驗分別制備磁性納米顆粒Fe3O4及共價有機骨架材料COF-SCU1,再將酸化處理的Fe3O4與COF-SCU1通過靜電吸附作用制備磁性共價有機骨架材料Fe3O4@COF-SCU1,并基于Fe3O4@COF-SCU1擬建立磁性固相萃取—高效液相色譜/紫外法分析大紅袍茶湯中的8種PAHs,以期為檢測環境和食品中的PAHs提供相關的理論依據。
1 材料與方法
1.1 材料與儀器
1.1.1 材料與試劑
1,3,5-苯三甲酰氯:純度≥98%,上海阿拉丁生化科技股份有限公司;
甲醇、乙腈:色譜純,國藥集團上海化學試劑有限公司;
對苯二胺、三氯化鐵、乙酸鈉、乙酸乙酯、乙醇、乙二醇、丙酮、二氯甲烷:分析純,國藥集團上海化學試劑有限公司;
芴(Flu)、菲(Phe)、蒽(Ant)、熒蒽(Flt)、芘(Pyr)、苯并(a)蒽(BaA)、苯并(b)熒蒽(BbF)、苯并(a)芘(BaP)標準品:純度≥99%,上海阿拉丁生化科技股份有限公司;
茶葉:大紅袍,福建省武夷山市。
1.1.2 主要儀器設備
高效液相色譜儀:LC-20A型,配有紫外檢測器,島津-GL(上海)商貿有限公司;
電子天平:ME204/02型,梅特勒—托利多儀器(上海)有限公司;
超聲波清洗器:KQ5200E型,昆明市超聲儀器有限公司;
超純水系統:Milli-Q IQ7000型,美國Merck公司;
氮吹儀:MG-2200型,上海虔鈞科學儀器有限公司;
X射線粉末衍射(XRD):D8 Advance型,德國Bruker公司;
超導量子干涉裝置磁力計(SQUID):PPMS-9型,美國Quantum Design公司;
物理吸附分析儀:ASAP 2020型,美國Micromeritics公司;
傅里葉變換紅外(FT-IR)光譜儀:Nicolet 5700型,美國Nicolet公司;
透射電子顯微鏡(TEM):FEI Tecnai G20型,美國FEI公司;
掃描電子顯微鏡(SEM):FEI Inspect QUANTA 430型,美國FEI公司。
1.2 方法
1.2.1 Fe3O4@COF-SCU1的制備
(1) Fe3O4合成:根據文獻[31]。將2.7 g三氯化鐵和7.2 g乙酸鈉溶解在100 mL乙二醇中,并將混合液置于反應釜中在200 ℃下反應8 h。反應結束后,用乙醇和超純水清洗干凈,于50 ℃真空干燥箱中干燥備用。
(2) COF-SCU1合成:根據文獻[17]。將1.06 g 1,3,5-苯三甲酰氯溶解在30 mL乙酸乙酯中。將0.32 g對苯二胺溶解于15 mL乙酸乙酯,在0 ℃冰水浴條件下逐滴滴入1,3,5-苯三甲酰氯溶液中并在室溫下攪拌24 h,反應結束后將所得的產物用乙醇和超純水清洗干凈,于50 ℃真空干燥箱中干燥備用。
(3) 制備Fe3O4@COF-SCU1:將2 g的Fe3O4溶解于0.1 mol/L的HCl溶液中,超聲處理10 min,然后用超純水清洗干凈,并分散于20 mL超純水中;將2 g的COF-SCU1分散于20 mL的超純水中,超聲處理10 min。將上述兩種溶液混合,渦旋1 min,即可得Fe3O4@COF-SCU1。
1.2.2 色譜條件 色譜柱:Agilent C18(4.6 mm×150 mm,5 μm),柱溫30 ℃,進樣體積20 μL,流速1 min/mL。流動相:A為水,B為乙腈;梯度洗脫程序:0~5 min,60% B;5~11 min,60%~100% B;11~16 min,100% B;16~21 min,100%~60% B;21~25 min,60% B。PAHs的檢測波長:Flu 210 nm,Phe 251 nm,Ant 251 nm,Flt 232 nm,Pyr 238 nm,BaA 287 nm,BbF 258 nm,BaP 295 nm。
1.2.3 混合標準溶液的配制 分別稱取Flu、Phe、Ant、Flt、Pyr、BaA、BbF、BaP標準品,精確至0.000 1 g,用乙腈配制成1 000 μg/mL 的儲備液。將8種標準儲備液混合配制成200 μg/mL的混合標準溶液。用水逐級稀釋質量濃度分別為4.00,2.00,1.00,0.50,0.25,0.10 μg/mL的標準溶液。將上述溶液置于4 ℃的冰箱中保存。
1.2.4 樣品前處理 0.5 g大紅袍茶葉加入120 mL開水沖泡3 min,茶湯用0.45 μm濾膜過濾,冷卻至室溫備用。
1.2.5 磁性固相萃取過程 萃取試驗過程具體步驟如圖1所示,將10 mg磁性吸附劑材料加入40 mL PAHs加標水樣中,渦旋3 min使分析物與磁性材料充分接觸。通過磁鐵使吸附劑從水樣中分離并去除上清液。加入2 mL二氯甲烷—乙腈(體積比1∶4)渦旋4 min,收集洗脫液并用溫和的氮氣吹至200 μL,用流動相定容至500 μL,經 0.22 μm微孔濾膜過濾,取20 μL于HPLC分析。

圖1 Fe3O4@COF-SCU1的制備及MSPE-HPLC-UV法檢測PAHs示意圖
Figure 1 Schematic diagram of the preparation of Fe3O4@COF-SCU1 and the detection of PAHs by MSPE-HPLC-UV
2 結果與討論
2.1 Fe3O4@COF-SCU1的表征
2.1.1 形貌表征 圖2為SEM和TEM表征結果,揭示了材料的微觀形態。如圖2(a)所示,COF-SCU1為聚集狀態,是由于大量的COF-SCU1團聚形成粒徑約為200 nm 的小球形顆粒。從圖2(b)可以觀察到COF-SCU1黏附在Fe3O4周圍和表面。從圖2(d)可以觀察到Fe3O4周圍分布的COF-SCU1,進一步說明了COF-SCU1已經成功修飾在Fe3O4表面。
2.1.2 磁性能(VSM)分析 從磁滯曲線(圖3)可知,Fe3O4和Fe3O4@COF-SCU1具有超順磁性,Fe3O4飽和磁化值高達79.8 emu/g,Fe3O4@COF-SCU1飽和磁化值為55.4 emu/g,這種高飽和磁性可賦予其對磁鐵的快速響應,利于收集和分離。
2.1.3 紅外光譜(FTIR)分析 為了考察所制備材料的化學基團,分別對Fe3O4、COF-SCU1和Fe3O4@COF-SCU1進行了FTIR表征見圖4。從COF-SCU1譜圖可以觀察到在3 378 cm-1處有很強的吸收峰,歸屬于—OH或—NH鍵的伸縮振動;在2 923 cm-1的峰為苯環C—H的伸縮振動峰;在1 713 cm-1的峰為苯環的特征吸收峰;在1 658 cm-1的峰為羧酸和酰胺C═O鍵的吸收峰;從Fe3O4譜圖可以觀察到578 cm-1處有強吸收峰,歸屬于Fe—O鍵伸縮振動引起的特征吸收峰,說明存在Fe3O4。從Fe3O4@COF-SCU1的FTIR圖中清晰可見Fe3O4和COF-SCU1的特征峰,結果表明了Fe3O4@COF-SCU1為Fe3O4和COF-SCU1組成的復合材料。

圖2 Fe3O4、COF-SCU1和Fe3O4@COF-SCU1的SEM圖和TEM圖
Figure 2 SEM images and TEM images of Fe3O4, COF-SCU1 and Fe3O4@COF-SCU1

圖3 Fe3O4和Fe3O4@COF-SCU1的磁滯曲線
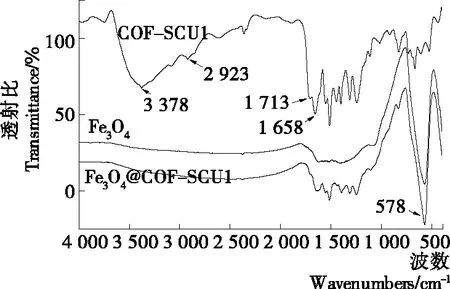
圖4 Fe3O4、COF-SCU1和Fe3O4@COF-SCU1的FTIR圖
2.1.4 X射線衍射(XRD)分析 采用XRD考察了Fe3O4、COF-SCU1和Fe3O4@COF-SCU1的晶體結構,結果見圖5。Fe3O4分別在30.24°,35.48°,43.11°,57.00°,62.66°處有明顯的特征衍射峰,與文獻[31]相一致。COF-SCU1在26°處有較寬的衍射峰,說明其為無定型結構。從Fe3O4@COF-SCU1的XRD圖可以觀察到Fe3O4和COF-SCU1的特征峰均有出現,再次表明了Fe3O4@COF-SCU1為Fe3O4和COF-SCU1組成的復合材料。
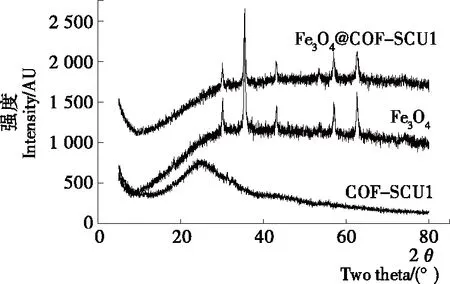
圖5 Fe3O4、COF-SCU1和Fe3O4@COF-SCU 1的XRD圖
2.1.5 比表面積(BET)氮氣吸附脫附表征 通過氮氣等溫吸脫附,得Fe3O4的BET比表面積和孔體積分別為25.44 m2/g 和0.077 cm3/g。COF-SCU1的BET比表面積和孔體積分別為60.59 m2/g和0.47 cm3/g。
2.2 MSPE條件優化
2.2.1 Fe3O4與COF-SCU1配比的影響 Fe3O4@COF-SCU1主要通過疏水相互作用,π-π堆積作用力來萃取分析物,為獲得最佳的萃取效果,分別制備了Fe3O4和COF-SCU1的質量比為5∶5,6∶4,7∶3,8∶2的吸附劑。如圖6(a)所示,當質量比為5∶5時,觀察到8種PAHs有較佳的回收率,且均在61%以上,其中BbF和BaP的回收率超過了90%。因此,后續試驗使用的是基于Fe3O4和COF-SCU1的質量比為5∶5的吸附劑。
2.2.2 離子強度的影響 為了研究離子強度對萃取分析物的影響,使用NaCl來調節溶液的鹽溶度。如圖6(b)所示,當NaCl濃度從0.02 mol/L增加到0.06 mol/L時,分析物的回收率基本保持不變。而繼續添加鹽時,回收率均有所下降。這可能是添加的鹽增加了溶液的黏度,影響了PAHs在吸附劑上的吸附動力學,因此后續試驗中不添加NaCl。
2.2.3 吸附程序的優化
(1) 吸附劑用量對萃取效果的影響:考察了不同用量(5,10,15,20,25 mg)的吸附劑對萃取效率的影響。如圖6(c) 所示,當吸附劑從5 mg增加到10 mg,PAHs回收率均有著顯著的提高。其后隨著吸附劑用量的增加,回收率基本保持恒定。因此,后續試驗中吸附劑用量選擇10 mg。

圖6 磁性固相萃取條件優化
(2) 萃取時間對萃取效果的影響:如圖6(d)所示,萃取3 min時8種PAHs即可達到吸附平衡。因此,在后續試驗中萃取時間選擇3 min。
2.2.4 洗脫程序的優化 選取甲醇、乙醇、丙酮、乙腈以及二氯甲烷—乙腈(體積比1∶4)5種洗脫劑進行了探索。從圖6(e)可以觀察到,二氯甲烷—乙腈(體積比1∶4)解析效果最好。因此,進一步優化了洗脫時間和二氯甲烷—乙腈(體積比1∶4)的用量,結果如圖6(f)和(g)所示。最終選擇2 mL的二氯甲烷—乙腈(體積比1∶4)作為洗脫溶劑,洗脫時間為4 min。
2.3 重復利用性
為考察材料的重復利用性,每次MSPE過程后,分別用1 mL的甲醇和水清洗吸附劑,再進行試驗。經過5次試驗,PAHs的回收率沒有明顯變化,說明Fe3O4@COF-SCU1具有良好的穩定性。
2.4 方法的線性范圍與檢出限
通過使用優化的MSPE條件確定方法的線性范圍,靈敏度,驗證方法的分析性能。配制一系列濃度的標準溶液,在最佳萃取條件下,經MSPE后進行HPLC-UV分析,如表1所示,8種PAHs均得到良好的線性關系,相關系數≥0.998 7。檢測限(LODs,S/N=3)和定量限(LOQs,S/N=10)分別為0.10~0.40,0.33~1.34 ng/mL。
2.5 實際樣品的分析
將建立的方法應用于大紅袍茶湯中8種PAHs的分析檢測。結果表明,大紅袍茶湯中的Ant的含量為0.471 2 ng/mL,其余的PAHs未檢出。分別添加濃度為2,10,20 ng/mL的PAHs混合標準溶液進行加標回收試驗,重復3次。如表2所示,8種PAHs的平均回收率在74%~106%,相對標準偏差為1.20%~8.50%。圖7為大紅袍茶湯中分別加入濃度為2,10 ng/mL的PAHs混合標準溶液前后對比的色譜圖。上述試驗結果表明:該方法滿足分析檢測的要求,可用于檢測低濃度的PAHs。
3 結論
通過簡易的方法成功制備了Fe3O4@COF-SCU1并將其作為吸附劑,建立MSPE-HPLC-UV法檢測大紅袍茶湯中PAHs的分析方法。該方法操作簡單,快速準確,充分利用了COF-SCU1的吸附能力以及MSPE的優勢,避免了過濾等繁瑣操作,且吸附劑可以快速方便地回收再利用,在同時富集萃取多種PAHs方面具有很好的應用前景。此外,研究出該技術的自動化萃取裝置,實現在線磁性萃取進行實時檢測分析,是該技術的進一步發展方向。

表1 方法的線性方程、線性范圍、相關系數、檢出限和定量限
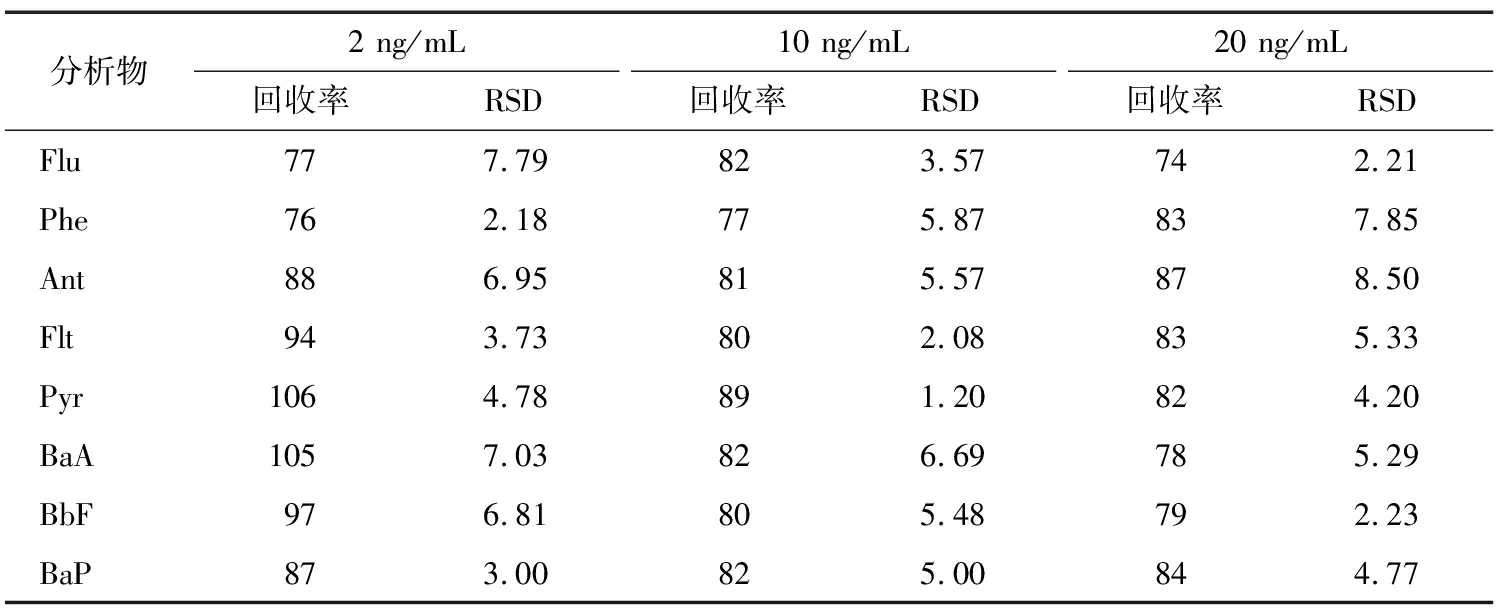
表2 大紅袍樣品中8種PAHs的加標回收率和相對標準偏差
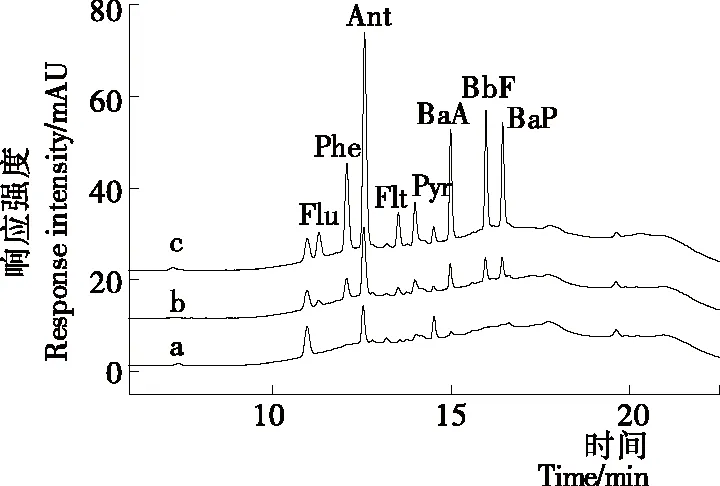
a. 沒有加標 b. 加標2 ng/mL c. 加標10 ng/mL
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
