乳鐵蛋白-EGCG共價復合物對魚油乳液穩定性和消化特性的影響
2020-01-07 03:18:02劉潤華梁秀萍閆曉佳劉夫國
食品科學 2019年24期
劉潤華,梁秀萍,閆曉佳,劉夫國,*
(1.食品營養與人類健康北京高精尖創新中心,北京工商大學,北京 100048;2.莫北農林科技大學食品科學與工程學院,陜莫 楊凌 712000)
食品生物活性成分,例如香料、色素、抗氧化劑、維生素、礦物質、魚油等,由于其與食品基質不相容以及在加工和貯存過程中穩定性差,不能被簡單地添加到食品和飲料中[1]。將生物活性成分包埋在膠體遞送系統,可以有效地克服以上問題。基于乳液的遞送系統特別適合于包埋、保護和控制釋放親脂性生物活性成分[2]。在乳液體系中,生物活性成分溶解在脂質相中,其通過均質轉化為乳化劑包埋的脂質液滴。選擇合適的乳化劑是決定乳液遞送系統理化性質和功能性能的最重要因素之一[3]。
傳統上,食品乳液通過單一的食品級乳化劑如小分子表面活性劑、磷脂、蛋白質或多糖穩定[4]。然而,開發基于不同分子(例如蛋白質-多糖、蛋白質-多酚或多糖-多酚)形成共價復合物或物理混合物作為多功能新型乳化劑的研究近年來引起了人們極大的興趣[5-7]。前期研究表明,蛋白質-多糖和蛋白質-多酚共價復合物具有優良的抗氧化、乳化和空間穩定性[8-9]。此外,在油滴界面上形成的蛋白質-多糖靜電復合物能夠用于改善乳液對環境應力的穩定性,并控制它們的包埋和釋放特性[10-11]。采用這些方法改變界面層的厚度、電荷和化學組成,可以設計具有增強諸如貯藏穩定性、化學穩定性的食品級乳液。
乳鐵蛋白(lactoferrin,LF)是一種鐵結合糖蛋白,含有約680 個氨基酸殘基,分子質量約80 kDa[12]。因其較好的表面活性,牛LF被用作模型球蛋白組裝界面層。同時,LF具有潛在的健康益處,例如抗癌、抗氧化、抗炎和抗微生物活性。表沒食子兒茶素沒食子酸酯(epigallocatechin gallate,EGCG)是從綠茶中提取分離得到的一種天然化合物,因其良好的發全性、突出的抗氧化活性和廣泛的生理活性而被普遍應用在食品工業領域中。前期研究表明,通過堿處理制備的LF-EGCG共價復合物由于具有抗氧化界面可以有效地抑制β-胡蘿卜素降解[13]。因此,推測EGCG共價結合LF能夠賦予LF較高的抗氧化活性。本研究通過測定LF-EGCG共價復合物和物理混合物穩定乳液的黏度、硫代巴比妥酸反應物(thiobarbituric acid reactive substance,TBARS)值和體外消化行為,系統研究乳液界面組成和結構對魚油乳液穩定性的影響,以期抑制魚油的氧化降解,延緩消化過程中脂肪酸的釋放,進而擴展魚油在食品工業中的應用。
1 材料與方法
1.1 材料與試劑
LF(純度>98%) 新莫蘭莫部乳業公司;EGCG(純度>99.5%) 北京北實縱橫科技發展有限公司;Ropufa 30 n-3魚油 瑞士DSM公司;異硫氰酸熒光素、尼羅紅染料、脂肪酶(來源于豬的胰臟,Type II)、豬胃蛋白酶、膽汁提取物、黏液素 美國Sigma公司;透析袋(截留分子質量12~14 kDa) 北京拜爾迪生物技術有限公司。
1.2 儀器與設備
BS224S電子天平 賽多利斯科學儀器有限公司;S20精密pH計 瑞士梅特勒-托利多儀器有限公司;ALPHA1-4/2-4冷凍干燥機 德國Christ有限公司;Ultrospec 3000 pro紫外分光光度計 英國Biochrom公司;PureNano雙通道高壓微射流 美國Microfluidics公司;Kinexus動態剪切流變儀、Nano-ZS90激光粒度儀 英國馬爾文公司;D-Eclipse C1 80i激光共聚焦顯微鏡 日本尼康公司;835 Titrando自動滴定儀瑞士萬通公司。
1.3 方法
1.3.1 共價復合物、物理混合物的制備
參考Rawel等[14]的方法合成LF-EGCG共價復合物。分別將1 g LF和0.2 g EGCG溶解在50 mL去離子水中并調至pH 9.0(質量濃度分別為2 g/100 mL和0.4 g/100 mL),攪拌過夜。將兩種溶液在連續攪拌條件下以1∶1的比例混合,將該混合物暴露于空氣24 h后采用去離子水透析48 h,產物每隔6 h換水一次以確保未反應的游離多酚完全透析出去。之后采用冷凍干燥裝置凍干,得到固體樣品。
物理混合物的制備,將LF、EGCG溶液調至pH 7.0,采用與上述相同的方法制備。
筆者前期對得到的產物進行凝膠電泳表征[13],結果表明本實驗可成功制備得到LF-EGCG共價復合物。
1.3.2 魚油納米乳液的制備
采用雙通道動態高壓微射流方法制備魚油水包油乳液。保持乳化劑與油的比例為1∶10,制備具有含油相體積分數10%的乳液,水相為含有LF、LF-EGCG共價復合物或LF-EGCG物理混合物的pH 7.0的水溶液。將水相和油相倒入兩個不同的玻璃杯中。然后通過空氣驅動的高壓微射流在13 kpsi的壓力下迫使油相和水相形成精細乳液。使用微流化器調節油相(fO)和水相(fW)的流速(mL/min)控制乳液中油的最終濃度。微流化器的總流量為500 mL/min。根據油相和水相的體積、油相(923 kg/m3)和水相的密度確定流速[15]。
1.3.3 粒徑及電位測定
1.3.3.1 粒徑測定
采用Nano-ZS90激光粒度儀測定乳液的粒徑。同時用多分散系數表示粒徑分布。為避免多重光散射對測量的影響,所有樣品在分析前用去離子水稀釋500 倍。每個樣品分析重復3 次,結果以平均值表示。
1.3.3.2 Zeta電位測定
采用Nano-ZS90激光粒度儀測定三元復合物溶液的Zeta電位。為減小多重散射對測量的誤差,實驗中所有樣品在分析測試前用去離子水稀釋500 倍。每個樣品分析重復3 次,結果以平均值表示。
1.3.4 乳液流變性質測定
參考Qiu Chaoying等[16]的方法采用動態剪切流變儀對魚油乳液的流變性質進行測定。測試夾具為CP4/40,選擇板狀測量池,將大約1.5 mL樣品置于錐體和板之間(間距1.0 mm,錐角4°),平衡5 min,加熱使其達到測量溫度。使用恒定的剪切速率(10 s-1)和不同的剪切速率(0.01~100 s-1)測量乳液的表觀黏度,通過流變儀提供的rSpace軟件進行控制和數據采集。
1.3.5 氧化指標測定
將魚油乳液裝于10 mL離心管中,在55 ℃的恒溫室中避光保存15 d,每隔3 d測量一次TBARS值。具體測定方法如下:將1 mL乳液與2 mL TBA試劑(體積分數15%三氯乙酸、0.25 mol/L HCl與體積分數2% TBA溶于乙醇)混合,放在帶螺帽的玻璃試管中,渦旋,于沸水中加熱15 min,移至室溫水中冷卻10 min,1 000×g離心15 min,放置10 min后,取上清液于532 nm波長處測定吸光度,TBARS濃度用1,1,3,3-氯化四乙銨所作標準校正曲線測定。所有分析均做3 組平行。
1.3.6 體外胃腸道消化模型的構建
參考Salvia-Trujillo等[17]的方法,采用由口腔、胃和小腸相組成的體外胃腸道模型測定所攝取樣品的消化特性。
1.3.6.1 口腔階段
根據Sarkar等[18]的方法制備含有黏蛋白和鹽的模擬唾液。將5 mL乳液與5 mL模擬唾液等份混合制備體積分數為1.25%油的最終混合物。將混合物的pH值調節至6.8并在37 ℃孵育10 min(100 r/min)。通常食物在口腔中停留的時間較短,設置10 min消化時間主要是基于樣品處理標準化的考慮,使每個樣品具有相同的處理條件。
1.3.6.2 胃消化階段
在37 ℃水浴條件下將20 mL從口腔階段產生的樣品與20 mL含有0.003 2 g/mL胃蛋白酶模擬液進行混合。將pH值調節至2.5并將樣品在100 r/min的孵育搖床中保持2 h(溫度37 ℃),以模擬胃液消化。
1.3.6.3 腸消化階段
使用自動滴定儀裝置模擬小腸階段的消化。將30 mL樣品調節至pH 7.0并置于溫控室(37 ℃)中。然后,將4 mL膽汁提取物(46.87 mg/mL)和1 mL溶于磷酸鹽緩沖液(5 mmol/L,pH 7.0)中的氯化鈣(110 mg/mL)溶液加入到樣品中。之后加入新制備的脂肪酶分散液(0.06 g溶解在2.5 mL pH 7.0的磷酸鹽緩沖液中)。檢測混合物的pH值并滴定至pH 7.0,記錄2 h期間從脂質消化釋放的游離脂肪酸(free fatty acid,FFA)所需NaOH溶液(0.05 mol/L)的體積,在時間t時FFA釋放率按下式計算:

式中:Vt(NaOH)為中和在消化時間t產生的FFA所需氫氧化鈉溶液的體積/L;C(NaOH)為滴定試樣的氫氧化鈉溶液的濃度(0.25 mol/L);Moil為魚油的分子質量(868 g/mol);moil為脂質最初存在于反應容器的總質量/g。
1.3.7 激光共聚焦顯微鏡測定
為觀察乳液在模擬胃腸道不同階段中的結構變化,利用共聚焦熒光掃描顯微鏡觀察乳液的微觀結構,采用油浸物鏡(×60)進行觀察。分析前,移取2 mL樣品于試管中,加入0.1 mL尼羅紅溶液(1 mg/mL,溶劑乙醇)和熒光素硫氰酸異構體I溶液(溶于二甲亞砜,1 mg/mL),并充分混合以確保均勻溶解。尼羅紅的激發和發射波長分別543 nm和605 nm,對于異硫氰酸熒光素分別設置為488 nm和515 nm。將混合物滴在載玻片上,在顯微鏡下觀察。采用尼康圖像處理軟件拍攝和分析乳液的微觀結構。
1.4 數據處理與分析
所有實驗至少進行3 次,所有數據為3 次測定的平均值。采用SPSS 17.0程序進行單因素方差分析,實驗結果以±s表示。采用Duncan檢驗進行數據比較和方差分析,P<0.05,差異顯著。
2 結果與分析
2.1 LF-EGCG共價復合物對魚油乳液流變性質的影響
由圖1a可以看出,在一定的剪切速率下,LF-EGCG共價復合物魚油乳液的黏度較高,這表明該乳液可能具有較高的物理穩定性。EGCG與LF共價復合后,LF的分子質量增加[19],根據鏈纏結理論[20],分子質量越大,分子鏈纏結越嚴重,使流動阻力變大,黏度升高。圖1b描述了不同乳液樣品的表觀黏度隨剪切速率變化而發生的變化。在整個剪切速率范圍內,LF和物理混合物樣品在較低的剪切速率下,其黏度逐漸降低,在較高的剪切速率下其黏度變化趨勢不明顯,乳液表觀黏度基本保持穩定,呈現牛頓流體行為。對于共價復合物樣品,其黏度在整個剪切速率范圍內隨剪切速率的增加而逐漸降低,呈現剪切變稀的特性。這主要是由于乳液內部單元的取方隨著剪切速率的增加與剪切方方一致或者平行,從而導致了黏度下降[21]。根據Stockes規律,顆粒黏度越大,沉淀速度越慢,乳液也越穩定。因此,基于幾種乳液樣品不同的流變學性質,其理化穩定性可能存在一定的差異。

圖1 LF、共價復合物和物理混合物穩定的魚油乳液在10 s-1剪切速率下的黏度(a)和流動圖(表觀黏度對剪切速率曲線)(b)Fig. 1 Apparent shear viscosity (at 10 s-1 shear rate) and fl owability pro files (shear viscosity versus shear rate) of fish oil emulsions stabilized by LF, the conjugate or the mixture
2.2 LF-EGCG共價復合物對魚油乳液外觀和氧化穩定性的影響
記錄了乳液樣品在55 ℃的恒溫條件下避光保存15 d前后的外觀形態(圖2a),同時采用TBARS方法檢測了油脂次級氧化產物的生成情況(圖2b),TBARS主要來源于脂質氫過氧化物的分解。如圖2a1所示,新鮮制備的不同魚油乳液的外觀呈現微小差異,這主要是由于在堿性條件下,EGCG與LF反應后,生成褐色共價復合物,導致最終產品呈現輕微的紅棕色。新鮮制備的乳液樣品流動性較好,然而,55 ℃恒溫貯藏15 d后,在LF和物理混合物的樣品中觀察到了凝膠現象(圖2a2),將試管倒置,液體仍能保留在試管的底部,說明這些樣品發生了絮凝,導致其流變性質發生明顯變化,由液體狀態變為類固體狀態。而對于共價復合物樣品,其外觀未發生明顯變化,貯藏15 d后仍能保持較好的流動性。
如圖2b所示,新鮮制備的魚油乳液中TBARS值小于1.0 nmol/g。在LF、LF-EGCG物理混合物和共價復合物穩定的魚油乳液中,其TBARS值分別為0.80、0.65 nmol/g和0.60 nmol/g,說明以物理復合或共價結合的形式加入EGCG均能使乳液具有較高的氧化穩定性。在貯藏過程中,乳液中脂質的TBARS值逐漸升高,在貯藏第15天時分別達到2.25、0.75 nmol/g和0.70 nmol/g。同一貯藏時間條件下,物理混合物和共價復合物的TBARS值相對較低,說明EGCG的存在能夠抑制脂質的氧化。研究表明,在乳液界面處,即脂滴和連續相的接觸區域,是油脂最容易氧化的區域[22]。本實驗中物理混合物和共價復合物穩定的乳液的氧化穩定性較高,主要歸因于EGCG對油水界面處自由基和過渡金屬的清除和鈍化作用。EGCG可能起到斷鏈型抗氧化劑的作用,抑制氫過氧化物分解為烷氧基和過氧化自由基,以及這些自由基進攻不飽和脂肪酸形成新的自由基[23]。同時,本實驗中交聯狀態的EGCG可以增加界面層的厚度,游離狀態的EGCG能夠有效地交聯油水界面的分子,形成致密的吸附層,從而起到了延緩油脂氧化的作用。
總體來說,LF-EGCG共價復合物不僅能提高貯藏過程中乳液的物理穩定性,而且能較好地抑制脂質氧化,提高魚油乳液的化學穩定性。
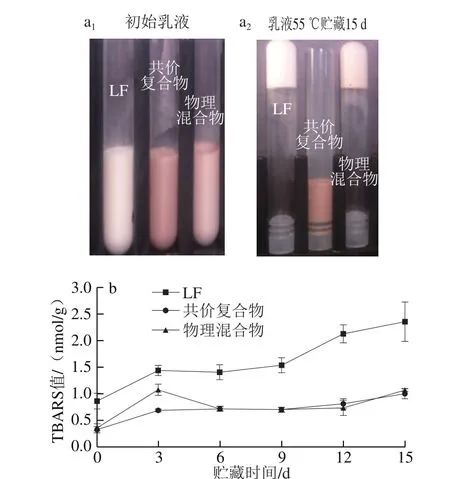
圖2 LF、LF-EGCG共價復合物和物理混合物穩定乳液的視覺表觀(a)和TBARS值(b)Fig. 2 Visual appearance (a) and TBARS values (b) of emulsions stabilized by LF, the conjugate or the mixture
2.3 消化過程中魚油乳液的粒徑、電位及微觀結構
模擬消化有助于進一步了解乳液在機體內的生物利用率、脂肪吸回等過程[24-25],促進特定屬性的功能食品設計。為探明LF、LF-EGCG物理混合物和共價復合物在體內的消化差異,進一步測定了不同乳液樣品在體外胃腸道模型中不同階段的粒徑、電位、粒徑分布和微觀結構(圖3~5)的變化。

圖3 不同魚油乳液在體外消化過程中的平均液滴大小(a)和Zeta電位(b)Fig. 3 Mean droplet size (a) and zeta-potential (b) of fish oil emulsions during in vitro gastrointestinal digestion

圖4 不同魚油乳液在體外消化過程中的粒徑分布Fig. 4 Particle size distribution of fish oil emulsions during in vitro gastrointestinal digestion
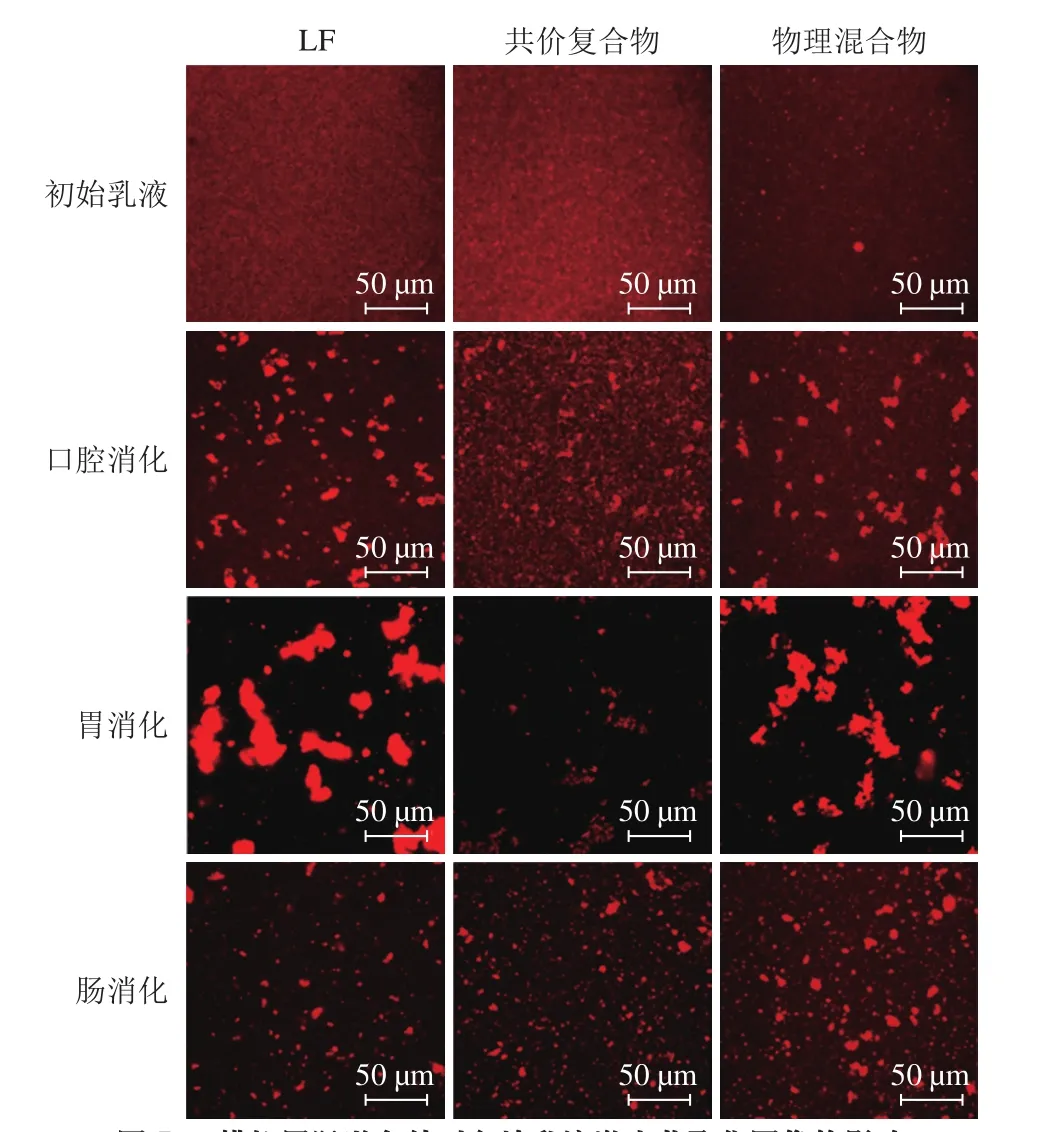
圖5 模擬胃腸道條件對魚油乳液激光共聚焦圖像的影響Fig. 5 Inf l uence of simulated gastrointestinal conditions on confocal images of fish oil emulsions
2.3.1 初始階段
由圖3可知,新鮮制備的乳液樣品的平均粒徑差異并不顯著。此外,激光共聚焦顯微鏡圖像顯示不同乳液樣品的液滴呈現均勻分布(圖5),說明3 種乳化劑在均質過程中產生的小液滴能夠抑制聚集。3 種乳液樣品的粒徑均呈單峰分布,表明在3 種新鮮制備的樣品中,LF及其復合物樣品均能發揮較好的乳化活性。
2.3.2 口腔階段
當乳液樣品暴露于口腔環境后,乳液的平均粒徑均呈現一定程度的增加,單獨蛋白質穩定的樣品粒徑增加較多(圖3a),粒徑分布顯示LF樣品變為雙峰,而物理混合物和共價復合物樣品仍為單峰分布(圖4)。微觀圖像結果表明粒徑增加是大量液滴聚集的結果(圖5)。這種現象歸因于乳滴與唾液中的黏蛋白發生了靜電或疏水相互作用,黏蛋白能夠誘導蛋白質穩定的乳液產生橋連或耗盡絮凝[26-27]。而LF-EGCG物理混合物或共價復合物能形成較厚的界面層,在一定程度上減弱乳滴與黏蛋白之間的相互作用。
2.3.3 胃消化階段
當乳液從口腔條件轉移至胃部階段時,所有乳液平均粒徑均顯著增加(圖3a)。這主要由于當乳滴分散于胃液中時,環境的pH值、離子強度和酶活性發生改變。胃中pH值較低,導致乳液的電位絕對值下降(圖3b)。因此,脂質液滴之間的靜電斥力減小,促進絮凝的產生。同時,胃液中較高的離子強度也會降低體系中的靜電作用,促進液滴聚集。另一方面,在胃蛋白酶的作用下,脂滴表面的蛋白質分子發生水解,同樣可以促進液滴聚集。微觀圖像顯示(圖5),由LF和LF-EGCG物理混合物穩定的脂滴含有較大的聚集體,而由共價復合物穩定的脂滴含有較小的聚集體,其分布相對均勻,這表明EGCG與LF共價結合可以改善脂滴在胃液條件下的聚集穩定性。這與蛋白質-多糖共價復合物可以改善乳液在胃腸道條件的聚集穩定性是一致的,其原因是共價復合物通過較強的空間排斥作用抑制了液滴之間的聚集[28-29]。
2.3.4 小腸消化階段
當乳液轉移至小腸環境時,與胃液的條件相比,所有乳液的平均粒徑均有所減小(圖3a),這主要由于乳液的聚集穩定性受pH值、離子強度和酶活性的影響。由圖4可知,所有乳液的電位絕對值均有所增加,因此脂滴之間的靜電排斥作用增加。同時,離子強度的改變、脂肪酶對脂滴的水解作用等也將影響脂滴的大小和存在狀態。微觀物理混合物樣品和共價復合物樣品的微結構之間存在明顯差異(圖5)。這些結果再次證明不同壁材的組成和相互作用可改變乳液的胃腸道消化性質。
2.4 LF-EGCG共價復合物對魚油乳液脂肪酸釋放率的影響
實驗采用pH值自動滴定儀測量了小腸條件下不同樣品脂肪消化速率和消化程度,通過記錄在pH值保持中性條件所消耗的NaOH溶液體積進一步計算出脂質水解產生的FFA[30]。由圖6可知,3 種乳液樣品在消化的最初幾分鐘內FFA釋放快速增加,在隨后消化過程中緩慢增加,表明脂酶分子可以快速吸附到油水界面,然后轉化三酰基甘油為FFA和單酰基甘油。不同乳液樣品的FFA釋放曲線存在微小差異。共價復合物樣品穩定的魚油乳液FFA釋放相對較慢,說明共價復合物形成的較厚的界面層在一定程度上可以抑制脂質水解。綜上所述,LF-EGCG共價復合物能在一定程度上延緩脂肪酸的釋放。
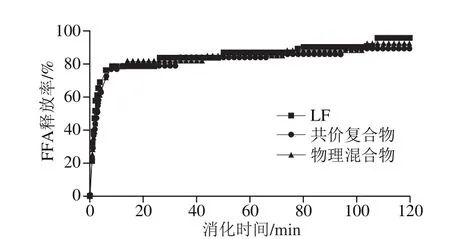
圖6 由LF、LF-EGCG共價復合物和物理混合物穩定的魚油乳液FFA釋放率Fig. 6 Percentage release of free fatty acids from fish oil emulsions stabilized by LF, the conjugate or the mixture
3 結 論
本研究發現通過堿法制備的LF-EGCG共價復合物對魚油乳液氧化穩定性及體外消化穩定性具有積極影響。在貯藏過程中,與LF和LF-EGCG物理混合物乳液相比,LF-EGCG共價復合物魚油乳液的黏度更高且具有更好的貯藏穩定性與氧化穩定性。在模擬體外消化過程中,LF-EGCG共價復合物能在一定程度上延緩脂肪酸的釋放。
