Swiprosin-1/EFhd2調(diào)控免疫炎癥反應(yīng)的研究進(jìn)展
2020-02-18 09:16:56馮美霞王志斌張立超
中國(guó)藥理學(xué)通報(bào) 2020年2期
馮美霞,蘇 笠,王志斌,張立超
(1. 上海市中醫(yī)醫(yī)院藥劑科,上海 200071;2. 上海大學(xué)轉(zhuǎn)化醫(yī)學(xué)研究院,上海 200444;3. 第二軍醫(yī)大學(xué)藥學(xué)系,上海 200433)
免疫炎癥反應(yīng)(immuno-inflammatory response)是通過細(xì)胞因子激活機(jī)體免疫細(xì)胞的活化,從而進(jìn)一步釋放細(xì)胞因子的過程。Swiprosin-1又稱EF-hand domain-containing protein D2(EFhd2),是一種保守的EF手性卷曲螺旋蛋白,主要在淋巴細(xì)胞和免疫系統(tǒng)中發(fā)揮作用[1]。目前研究發(fā)現(xiàn),EFhd2參與肥大細(xì)胞、巨噬細(xì)胞和B淋巴細(xì)胞等免疫細(xì)胞的炎癥因子分泌、遷移和凋亡,以及核轉(zhuǎn)錄因子κB(NF-κB)、Janus激酶/信號(hào)轉(zhuǎn)導(dǎo)和轉(zhuǎn)錄激活因子(JAK-STAT)、絲裂原活化蛋白激酶(MAPK)和鈣信號(hào)等炎癥信號(hào)通路的調(diào)節(jié),表明EFhd2具有廣泛的免疫調(diào)節(jié)潛能。本文主要綜述了EFhd2在調(diào)控免疫炎癥反應(yīng)中的研究進(jìn)展。
1 EFhd2的結(jié)構(gòu)及表達(dá)分布
EFhd2是一種由240個(gè)氨基酸組成的鈣銜接蛋白,分子量為27 ku(預(yù)測(cè)值)和33 ku(表觀值)。通過蛋白質(zhì)一級(jí)結(jié)構(gòu)分析顯示,EFhd2由4個(gè)推定的豆蔻酰化位點(diǎn)(脂質(zhì)修飾),3個(gè)SH3結(jié)構(gòu)域蛋白的結(jié)合位點(diǎn),2個(gè)功能性EF手性結(jié)構(gòu)域和C末端卷曲螺旋結(jié)構(gòu)域組成[1-2]。
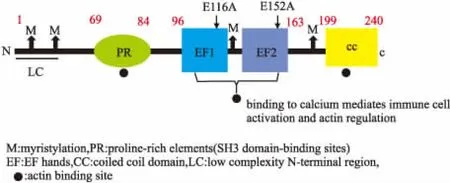
Fig 1 Protein primary structure patterns of EFhd2[2]
EFhd2最初在人的CD8+T淋巴細(xì)胞中被發(fā)現(xiàn)[1],隨后發(fā)現(xiàn)其在未成熟的B細(xì)胞、肥大細(xì)胞、自然殺傷細(xì)胞(NK細(xì)胞)、人外周血單核細(xì)胞(PBMC)等其它免疫細(xì)胞中也有表達(dá)[2-3]。并且,EFhd2廣泛表達(dá)于不同物種的組織和細(xì)胞中,在腦、肝臟、心臟、脾臟、肺、腎等組織中均有表達(dá)。研究還發(fā)現(xiàn),EFhd2在許多病理狀態(tài),如炎癥(急性-被動(dòng)皮膚過敏和慢性特異性皮炎)、神經(jīng)退行性疾病、精神分裂癥、膿毒癥、糖尿病腎病及腸球菌感染的男性生殖病等情況下均顯示表達(dá)增高[4],表明EFhd2與免疫和炎癥反應(yīng)具有一定相關(guān)性。
2 EFhd2在免疫炎癥反應(yīng)中的調(diào)節(jié)作用
2.1 EFhd2參與免疫細(xì)胞的激活目前研究發(fā)現(xiàn),從果蠅到高等生物,EFhd2在小鼠單核巨噬細(xì)胞RAW264、人PBMC、小膠質(zhì)細(xì)胞和NK樣細(xì)胞中均有表達(dá)。并且,EFhd2參與了T細(xì)胞晚期活化的蛋白激酶C(PKC)信號(hào)通路的表達(dá)調(diào)節(jié)和T細(xì)胞誘導(dǎo)的細(xì)胞毒性,在肥大細(xì)胞活化期間表達(dá)上調(diào);促進(jìn)巨噬細(xì)胞凋亡、遷移和炎癥因子的分泌;可作為B細(xì)胞脂筏(BCR)相關(guān)的銜接蛋白參與鈣離子信號(hào)的傳導(dǎo)等,這些結(jié)果表明,EFhd2可能作為免疫炎癥反應(yīng)調(diào)控中的潛在靶標(biāo)。
2.1.1在T細(xì)胞中的作用 作為免疫反應(yīng)的核心參與者,T細(xì)胞可通過與靶細(xì)胞特異性結(jié)合,破壞靶細(xì)胞膜直接殺傷靶細(xì)胞或釋放淋巴因子使免疫效應(yīng)擴(kuò)大和增強(qiáng)。基質(zhì)細(xì)胞衍生因子-1α(SDF-1α)是T細(xì)胞的一般趨化因子,并且SDF-1α受體CXCR4在大多數(shù)淋巴細(xì)胞均存在表達(dá)[5]。過表達(dá)EFhd2增強(qiáng)了SDF-1α介導(dǎo)Jurkat T細(xì)胞在纖粘蛋白上的細(xì)胞遷移[5]。但Kim等[6]發(fā)現(xiàn),EFhd2的過表達(dá)不參與T細(xì)胞抗原受體(T cell receptor,TCR)刺激或細(xì)胞內(nèi)信號(hào)激活誘導(dǎo)的T細(xì)胞活化,提示其不參與最初的T細(xì)胞活化;而使用抗CD3/28抗體刺激Jurkat T細(xì)胞6~12 h后,EFhd2 mRNA在T細(xì)胞活化期間表達(dá)明顯增加,并且其蛋白水平在PMA刺激下以時(shí)間依賴方式上調(diào),表明EFhd2可能參與T細(xì)胞晚期的PKC信號(hào)通路的活化[6]。
程序性細(xì)胞死亡蛋白-1(PD-1)是一種典型的共抑制受體,在免疫抑制中通過結(jié)合配體PD-LI或PD-L2激活T淋巴細(xì)胞。Michael等[7]發(fā)現(xiàn)EFhd2與免疫突觸中的PD-1共定位并且可作為PD-1信號(hào)轉(zhuǎn)導(dǎo)的重要介質(zhì)。通過小干擾RNA(siRNA)干擾人原代T細(xì)胞中的EFhd2,幾乎完全消除了PD-1對(duì)抗原受體下游IL-2分泌的抑制作用,而敲減EFhd2阻斷了PD-1誘導(dǎo)的細(xì)胞外調(diào)節(jié)蛋白激酶(ERK)去磷酸化。此外,將MC38鼠結(jié)腸癌細(xì)胞植入EFhd2敲除鼠皮下,與同窩野生型小鼠相比,敲除鼠的細(xì)胞毒性降低、細(xì)胞中溶解顆粒向免疫突觸極化受損以及腫瘤生長(zhǎng)速率增加,表明EFhd2對(duì)于T細(xì)胞誘導(dǎo)的細(xì)胞毒性也是必需的[7]。
2.1.2在肥大細(xì)胞中的作用 在體外培養(yǎng)的人肥大細(xì)胞HMC-1中,Kim等[8-9]發(fā)現(xiàn),佛波酯對(duì)EFhd2有瞬時(shí)誘導(dǎo)作用,過表達(dá)EFhd2能增強(qiáng)PKA/A23187刺激HMC-1分泌組胺和IL-8,增加IL-8和IL-3 mRNA的表達(dá),而敲減EFhd2可抑制IL-8和IL-3 mRNA表達(dá)。此外,他們還通過IgE和人血清白蛋白(DNP-HAS)刺激的FcεRI交聯(lián),以及被動(dòng)皮膚過敏反應(yīng)和特應(yīng)性皮炎的體內(nèi)組織模型發(fā)現(xiàn)RBL-2H3細(xì)胞中EFhd2的表達(dá)上調(diào)。這些結(jié)果證明了在炎癥反應(yīng)中,EFhd2可能作為細(xì)胞因子表達(dá)和肥大細(xì)胞活化的調(diào)節(jié)劑[8]。
2.1.3在巨噬細(xì)胞中的作用 巨噬細(xì)胞是抵抗入侵病原體的起始防御線之一,是具有誘導(dǎo)初級(jí)免疫應(yīng)答能力的抗原遞呈細(xì)胞。通過釋放各種細(xì)胞因子如腫瘤壞死因子-α(TNF-α),巨噬細(xì)胞可參與調(diào)節(jié)適應(yīng)性免疫應(yīng)答,在重組牛分枝桿菌株(BCG)對(duì)人單核細(xì)胞系THP-1細(xì)胞的刺激下,EFhd2與TNF-α、CD40等一起被明顯上調(diào),增強(qiáng)了免疫刺激活性,改善了THP-1細(xì)胞的抗原遞呈能力[10]。此外,THP-1細(xì)胞在脂多糖(LPS)處理下顯示EFhd2的上調(diào),而使用siRNA敲減THP-1中EFhd2的表達(dá)則會(huì)降低LPS處理后上清液中IL-6和TNF-α的濃度,表明EFhd2的敲除會(huì)損害LPS 誘導(dǎo)的免疫反應(yīng)[10]。在膿毒癥小鼠中,我們研究發(fā)現(xiàn),與野生小鼠來(lái)源的腹腔巨噬細(xì)胞相比,EFhd2敲除小鼠來(lái)源的巨噬細(xì)胞表達(dá)更少的HLA-DR,吞噬作用和細(xì)菌殺傷能力也受損;并且EFhd2敲除小鼠在LPS或盲腸結(jié)扎穿孔術(shù)(CLP)誘導(dǎo)的膿毒癥中表現(xiàn)出更高的死亡率,嚴(yán)重的器官功能障礙,表明EFhd2參與巨噬細(xì)胞免疫應(yīng)答調(diào)控[11]。
當(dāng)局部損傷或炎癥發(fā)生時(shí),源自炎癥病灶的促炎信號(hào)(如促炎細(xì)胞因子和微生物)通過誘導(dǎo)單核細(xì)胞從骨髓中遷移,循環(huán)單核細(xì)胞通過滾動(dòng)、黏附和細(xì)胞骨架驅(qū)動(dòng)的局部?jī)?nèi)皮細(xì)胞遷移而靶向炎癥位點(diǎn),參與多種器官和組織的修復(fù)[12]。在人骨髓間充質(zhì)干細(xì)胞(BMSCs)到成骨細(xì)胞的分化過程中,和使用NF-κB受體激活蛋白配體(RANK-L)刺激小鼠巨噬細(xì)胞系RAW264細(xì)胞分化為破骨細(xì)胞的過程中,EFhd2 mRNA的表達(dá)量均表現(xiàn)明顯增高[13],表明EFhd2可能與炎癥疾病中BMSCs的自噬相關(guān),參與骨代謝的調(diào)控。另一方面,在主動(dòng)脈斑塊中的巨噬細(xì)胞中EFhd2也呈現(xiàn)高表達(dá),并伴隨泡沫化程度的加重而表達(dá)增加。EFhd2敲除后巨噬細(xì)胞中泡沫化程度明顯減弱,促凋亡相關(guān)蛋白Bax、caspase-3、caspase-9以及炎癥因子IL-1β、TNF-α水平減少,而抑凋亡蛋白Bcl-2表達(dá)增加,表明巨噬細(xì)胞表達(dá)的EFhd2加速動(dòng)脈粥樣硬化的進(jìn)展[14]。
2.1.4在B細(xì)胞中的作用 B細(xì)胞最初來(lái)源于骨髓的多能干細(xì)胞,成熟B細(xì)胞經(jīng)外周血進(jìn)入脾臟、淋巴結(jié),在抗原刺激后分化增殖為質(zhì)細(xì)胞,繼而合成和分泌抗體,參與體液免疫的執(zhí)行。先前存在的大量研究已證明,EFhd2從祖B細(xì)胞到漿母細(xì)胞階段均有表達(dá),并在骨髓未成熟的B細(xì)胞中表達(dá)最高,但也表達(dá)于靜息和激活的脾臟B細(xì)胞及其他非淋巴組織[2]。
B細(xì)胞受體(BCR)與其他因素一起控制著抗原誘導(dǎo)的繼發(fā)淋巴器官中B淋巴細(xì)胞的陽(yáng)性選擇和穩(wěn)態(tài)。值得注意的是,EFhd2在小鼠WEHI231細(xì)胞中可作為響應(yīng)BCR刺激的脾酪氨酸激酶(Syk)活性的正性調(diào)節(jié)劑[3],并且EFhd2可作為與B細(xì)胞脂筏相關(guān)的小銜接蛋白參與鈣信號(hào)的傳導(dǎo),控制B細(xì)胞受體信號(hào)轉(zhuǎn)導(dǎo)。過表達(dá)EFhd2的WEHI231細(xì)胞中,抗凋亡蛋白Bcl-2(BH)同源蛋白Bcl-XL大約減少了3倍。相反地,EFhd2沉默細(xì)胞中Bcl-XL蛋白與對(duì)照組相比平均增加了3倍。這些結(jié)果表明EFhd2可通過與Bcl-XL的相互調(diào)節(jié)控制WEHI231細(xì)胞群的凈細(xì)胞生長(zhǎng)和細(xì)胞的自發(fā)凋亡[2]。
在C57BL/6小鼠中敲除EFhd2并未影響B(tài)細(xì)胞對(duì)胸腺非依賴性抗原,如Nitrophenol-Ficoll和Trinitrophenol-LPS的免疫應(yīng)答。與野生小鼠相比,EFhd2敲除增加了感染巴西線蟲小鼠Th2細(xì)胞介導(dǎo)的IgE和IgM的產(chǎn)生。使用綿羊紅細(xì)胞(SRBC)和蠕蟲免疫小鼠7 d后,EFhd2敲除小鼠生發(fā)中心B細(xì)胞和其它同型抗體明顯增加,表明EFhd2在生發(fā)中心依賴性的體液免疫中可作為負(fù)性調(diào)節(jié)因子[15]。
2.2 EFhd2參與調(diào)節(jié)細(xì)胞遷移細(xì)胞骨架是一種由蛋白亞基組成的纖維網(wǎng)狀結(jié)構(gòu),對(duì)于細(xì)胞中亞細(xì)胞器的空間排布、細(xì)胞遷移、細(xì)胞分裂及細(xì)胞凋亡起著重要作用并最終決定整體細(xì)胞形狀[16]。細(xì)胞遷移的過程可分為前緣板狀偽足的形成,新黏附的建立,肌動(dòng)蛋白、肌球蛋白收縮與基質(zhì)分離完成。而這些過程均依賴于肌動(dòng)蛋白骨架的聚合、解聚及重新組裝,并受Rho蛋白調(diào)節(jié)。Rho家族蛋白是細(xì)胞骨架重組的主要調(diào)節(jié)因子之一,在細(xì)胞的遷移中起著重要作用。例如Cdc42是細(xì)胞極性調(diào)節(jié)和絲狀偽足形成所必需的;Rac1是板狀偽足、膜褶皺的形成和細(xì)胞遷移所必需的;而RhoA是細(xì)胞內(nèi)信號(hào)轉(zhuǎn)導(dǎo)通路的關(guān)鍵分子,參與遷移細(xì)胞后緣的縮回。
在富含肌動(dòng)蛋白細(xì)胞骨架的區(qū)域如HMC-1細(xì)胞的突起和293T細(xì)胞的膜頂端脊中EFhd2高度集聚,并呈現(xiàn)出肌動(dòng)蛋白結(jié)合活性[6,9]。例如,在胚胎成肌細(xì)胞融合過程中,果蠅EFhd2經(jīng)常與F-肌動(dòng)蛋白灶重疊[17];在PKC激動(dòng)劑佛波酯和表皮生長(zhǎng)因子(EGF)刺激下,EFhd2伴隨F-actin在COS-7和B16F10細(xì)胞中重新定位[9,18];此外,在具有肌動(dòng)蛋白和肌動(dòng)蛋白結(jié)合蛋白(如α-輔肌動(dòng)蛋白、塑性蛋白和細(xì)絲蛋白)的NK樣細(xì)胞、肥大細(xì)胞和黑素瘤細(xì)胞的細(xì)胞骨架部分中均發(fā)現(xiàn)了EFhd2,提示其可通過肌動(dòng)蛋白重塑調(diào)節(jié)細(xì)胞活化[5,9,19]。
我們先前的研究發(fā)現(xiàn),EFhd2可通過直接促進(jìn)肌動(dòng)蛋白聚合參與LPS刺激的巨噬細(xì)胞遷移[19];過表達(dá)EFhd2可增強(qiáng)板狀偽足的形成及LPS刺激的巨噬細(xì)胞遷移,而敲除EFhd2抑制了LPS誘導(dǎo)的Rac1/Cdc42和磷酸化Rac1/Cdc42的表達(dá)變化[19]。雖然,EFhd2不促進(jìn)由肌動(dòng)蛋白相關(guān)蛋白Arp2/3復(fù)合體和WASP蛋白的VCA結(jié)構(gòu)域介導(dǎo)的肌動(dòng)蛋白聚合,但EFhd2在絲氨酸殘基183(Ser183)位點(diǎn)的磷酸化可通過抑制肌動(dòng)蛋白絲的聚集和調(diào)節(jié)F-肌動(dòng)蛋白對(duì)cofilin的可及性參與肌動(dòng)蛋白的解聚[18]。類似的,Huh等[4]發(fā)現(xiàn)EFhd2異位表達(dá)增強(qiáng)了與肌動(dòng)蛋白相關(guān)的突起如板狀偽足和膜褶皺的形成,并激活B16F10細(xì)胞中的Rac1和Cdc42活性。
EFhd2位于70~199個(gè)氨基酸之間含有3個(gè)肌動(dòng)蛋白結(jié)合位點(diǎn)(69~96,96~163和163~199)[19]。在EFhd2的N端區(qū)域(氨基酸1~69)和C端卷曲螺旋區(qū)域(氨基酸200~240)中鑒定出非常弱或沒有肌動(dòng)蛋白結(jié)合位點(diǎn),然而這些位點(diǎn)對(duì)于由EFhd2調(diào)節(jié)的肌動(dòng)蛋白親和力是必不可少的[17,19]。與對(duì)照相比,敲除EFhd2的EF手基序(D97~163,M2)后顯示出較少的肌動(dòng)蛋白成束活性,而卷曲螺旋結(jié)構(gòu)域(1~163,M1)的敲除消除了肌動(dòng)蛋白成束活性,顯示細(xì)胞質(zhì)區(qū)域細(xì)胞鋪展和板狀偽足形成明顯減少,表明EFhd2的卷曲螺旋結(jié)構(gòu)中富含賴氨酸的區(qū)域(218~240)對(duì)于肌動(dòng)蛋白成束活性是必不可缺的[6]。
3 EFhd2調(diào)節(jié)免疫炎癥反應(yīng)的信號(hào)通路
已知EFhd2可參與多種信號(hào)通路的調(diào)節(jié)。如HMC-1肥大細(xì)胞的PKC-βI/η[8]、Jurkat T細(xì)胞的NF-κB和PKC-θ[6]、WEHI231 B細(xì)胞的BCR[2]、破骨細(xì)胞樣細(xì)胞的核因子κB受體活化因子配體(RANKL)[10]、巨噬細(xì)胞中的JAK2/STAT1/3[11]、腎小球內(nèi)皮細(xì)胞中的PKC-β[20]和腎小球足細(xì)胞的MAPK信號(hào)通路[21]、Ca2+信號(hào)的轉(zhuǎn)導(dǎo)[17]等。其中NF-κB、JAK-STAT和MAPK作為最主要的三條炎癥信號(hào)通路,EFhd2均被報(bào)道參與其中。
3.1 參與NF-κB信號(hào)通路轉(zhuǎn)導(dǎo)NF-κB是一種重要的免疫炎癥信號(hào)通路,參與調(diào)控促炎介質(zhì)如細(xì)胞因子、趨化因子和粘附因子等基因的表達(dá)。值得注意的是,EFhd2在NF-κB活化中具有雙重作用。在BCR信號(hào)通路中作為NF-κB活化的負(fù)調(diào)節(jié)劑[2],而在PKC信號(hào)通路中作為NF-κB的正調(diào)節(jié)劑[8]。
Kim等[6]發(fā)現(xiàn)在佛波酯PMA/A23187刺激后,Jurkat T細(xì)胞中異位表達(dá)的EFhd2顯著增強(qiáng)了NF-κB的轉(zhuǎn)錄活性和IκB-α降解,以及促炎細(xì)胞因子的表達(dá);而在特異性NF-κB活化抑制劑咖啡酸苯乙酯(CAPE)的刺激下,阻斷了EFhd2表達(dá)上調(diào)。此外,Nomiyama等[10]通過NF-κB配體的受體激活劑RANKL和TNF-α以及巨噬細(xì)胞炎性蛋白-1α(MIP-1α)三者聯(lián)用處理小鼠巨噬細(xì)胞RAW264.7后也發(fā)現(xiàn),EFhd2基因的表達(dá)上調(diào),表明NF-κB信號(hào)傳導(dǎo)對(duì)于巨噬細(xì)胞中EFhd2的誘導(dǎo)至關(guān)重要。
相反,Avramidou等[2]通過BCR刺激未成熟小鼠B淋巴細(xì)胞WEHI231,發(fā)現(xiàn)沉默EFhd2誘導(dǎo)了強(qiáng)烈的IκB-α磷酸化和降解,但在對(duì)照細(xì)胞和異位表達(dá)EFhd2的細(xì)胞中并沒有觀察到此現(xiàn)象,表明EFhd2在經(jīng)典NF-κB信號(hào)通路的活化中可能起負(fù)性調(diào)節(jié)作用。
3.2 參與JAK-STAT和MAPK信號(hào)通路轉(zhuǎn)導(dǎo)JAK-STAT信號(hào)通路作為多種細(xì)胞因子和生長(zhǎng)因子在細(xì)胞內(nèi)傳遞信號(hào)的共同途徑,參與炎癥和類風(fēng)濕關(guān)節(jié)炎、銀屑病和炎癥性腸病等多種自身免疫疾病。IFN-γ與其細(xì)胞表面受體IFN-γR結(jié)合后可激活下游的JAK-STAT信號(hào)通路,繼而激活某些抗炎基因的表達(dá),參與啟動(dòng)和調(diào)控免疫炎癥[22]。
前期研究中,我們發(fā)現(xiàn)EFhd2敲除抑制了由IFN-γ激活的STAT1和STAT3磷酸化[11]。與LPS處理后的野生型相比,EFhd2-/-膿毒癥小鼠血清與T細(xì)胞中IFNγ水平減少,巨噬細(xì)胞中IFN-γR的表達(dá)水平和JAK-STAT通路的活化(JAK2,STAT1和STAT3磷酸化)也均明顯降低;相反,過表達(dá)EFhd2明顯增加了JAK2/STAT1/3的磷酸化[11]。另一方面,曹雪濤等[22]發(fā)現(xiàn)EFhd2與布魯頓酪氨酸蛋白激酶(BTK)磷酸化的Ⅰ型IFN受體的β亞基(IFN-γR2Y289)相互作用,促進(jìn)IFN-γR2的膜轉(zhuǎn)運(yùn),并形成功能性IFN-γR以介導(dǎo)IFN-γ信號(hào)轉(zhuǎn)導(dǎo),從而參與啟動(dòng)巨噬細(xì)胞對(duì)于胞內(nèi)細(xì)菌感染的有效先天性免疫反應(yīng)。并且,李斯特菌感染的EFhd2敲除鼠與同窩野生型小鼠相比,在腹腔巨噬細(xì)胞的細(xì)胞膜上顯示出IFN-γR2表達(dá)顯著降低,并且其TNF-α和IL-6 mRNA也遠(yuǎn)低于野生型[22]。以上結(jié)果表明,EFhd2不僅影響IFN-γR1的表達(dá),而且對(duì)于驅(qū)動(dòng)IFN-γR2膜轉(zhuǎn)運(yùn)也是不可或缺的。
另一方面,MAPK信號(hào)通路在細(xì)胞的增殖、分化和凋亡中也起著非常重要的作用。我們前期研究發(fā)現(xiàn),腎小球足細(xì)胞MPC-5表達(dá)EFhd2,使用鏈脲佐菌素和高葡萄糖處理后EFhd2表達(dá)增加;雖然敲除EFhd2與野生型對(duì)照組之間p38的磷酸化水平并沒有明顯差異,但p38的磷酸化水平在野生型糖尿病小鼠中卻是增加的;EFhd2敲除糖尿病小鼠和高糖處理的EFhd2敲減的MPC-5細(xì)胞中p38的磷酸化水平及足細(xì)胞凋亡均受到抑制;給予p38抑制劑SB203580減少了高糖誘導(dǎo)的細(xì)胞凋亡。這些結(jié)果表明EFhd2在糖尿病腎病早期可通過激活p38 MAPK信號(hào)通路促進(jìn)足細(xì)胞凋亡[21]。
3.3 參與Ca2+信號(hào)轉(zhuǎn)導(dǎo)Ca2+是絕大部分細(xì)胞必不可缺的第二信使,持續(xù)的Ca2+流入對(duì)于淋巴細(xì)胞的激活和適應(yīng)性免疫反應(yīng)均起著至關(guān)重要的作用。Ca2+信號(hào)轉(zhuǎn)導(dǎo)通常通過EF手型(EF-hand)家族的Ca2+結(jié)合蛋白參與執(zhí)行,如通過鈣調(diào)神經(jīng)磷酸酶激活NFAT的鈣調(diào)蛋白[17]。
EFhd2作為一種Ca2+銜接蛋白,可將Syk,SLP-65和PLCγ2結(jié)合在一起介導(dǎo)BCR誘導(dǎo)的鈣離子流動(dòng)[1-2,9]。在小鼠B細(xì)胞系WEHI213細(xì)胞中,過表達(dá)EFhd2明顯增強(qiáng)了IgM抗體刺激后BCR誘導(dǎo)的游離鈣離子濃度,而敲除EFhd2顯示低鈣離子濃度[2,23]。并且與野生型EFhd2相比,富含脯氨酸的區(qū)域(PR)、N-端無(wú)序區(qū)域(LC)和EF1缺失的突變體并未恢復(fù)BCR誘導(dǎo)的鈣離子流動(dòng),表明EFhd2的PR、LC和EF1區(qū)域?qū)τ贐CR誘導(dǎo)的鈣離子流動(dòng)是必不可缺的[2]。Kroczek等[3]發(fā)現(xiàn),EFhd2蛋白可通過負(fù)性調(diào)節(jié)NF-κB抗細(xì)胞凋亡途徑影響細(xì)胞存活和內(nèi)質(zhì)網(wǎng)儲(chǔ)存的鈣流出。然而,Morowski等[24]使用經(jīng)典血小板激動(dòng)劑如膠原相關(guān)肽(4 mg·L-1)和凝血酶(10-5U·L-1)刺激血小板15 min后,發(fā)現(xiàn)EFhd2敲除小鼠的血小板并未改變正常的Ca2+儲(chǔ)存和釋放,表明EFhd2對(duì)于血小板中的Ca2+信號(hào)傳導(dǎo)是非必需的。
通過構(gòu)建EFhd2蛋白N-末端聚丙氨酸區(qū)域、高度保守的C-末端卷曲螺旋區(qū)域和EF-手基序截短突變體并進(jìn)行鈣結(jié)合活性測(cè)定,F(xiàn)errer-Acosta等[25]發(fā)現(xiàn),EFhd2的鈣結(jié)合活性取決于兩個(gè)EF-手基序的完整性,N-末端和C-末端敲除增強(qiáng)了EFhd2的鈣結(jié)合能力。保守的谷氨酸殘基即E116和E152分別位于EFhd2的EF1和EF2的-Z位置。Hagen等[23]發(fā)現(xiàn),E116、E152單個(gè)突變使得鈣結(jié)合能力減半,而雙突變則鈣結(jié)合力消失,進(jìn)一步表明了EF1和EF2的保守殘基E116A和E152A是EFhd2鈣結(jié)合必不可少的。
4 結(jié)語(yǔ)
目前研究發(fā)現(xiàn),EFhd2與急性-被動(dòng)皮膚過敏和慢性特異性皮炎、膿毒癥、類風(fēng)濕關(guān)節(jié)炎、糖尿病腎病等自身免疫疾病相關(guān),并通過NF-κB、JAK-STAT、MAPK和Ca2+等多種信號(hào)通路參與炎癥細(xì)胞因子的激活,敲除EFhd2可抑制炎癥的發(fā)生,但也有部分報(bào)道認(rèn)為EFhd2可作為免疫炎癥的負(fù)性調(diào)節(jié)因子。總之,EFhd2在免疫和炎癥性疾病中的作用仍需要進(jìn)一步的研究,為免疫炎癥反應(yīng)的防治提供新的思路。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級(jí))(2025年8期)2025-08-18 00:00:00
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評(píng)價(jià)·高一版(2020年6期)2020-11-02 02:45:24
學(xué)苑創(chuàng)造·A版(2020年9期)2020-10-13 09:41:02
中國(guó)生殖健康(2019年3期)2019-02-01 06:12:26
小學(xué)生學(xué)習(xí)指導(dǎo)(低年級(jí))(2017年10期)2017-10-10 01:00:05
鑿巖機(jī)械氣動(dòng)工具(2016年3期)2016-03-01 04:00:25
海軍航空大學(xué)學(xué)報(bào)(2015年3期)2015-11-11 17:20:00
云南中醫(yī)學(xué)院學(xué)報(bào)(2014年3期)2014-07-31 18:57:34
七彩語(yǔ)文·畫刊(2012年3期)2012-04-29 00:00:00

- 中國(guó)藥理學(xué)通報(bào)的其它文章
- 濕潤(rùn)暴露療法/濕潤(rùn)燒傷膏對(duì)慢性創(chuàng)面組織中MMP-2和 MMP-9表達(dá)的影響
- 白藜蘆醇通過誘導(dǎo)自噬減少肥胖小鼠骨骼肌組織GDF8表達(dá)的研究
- 褪黑素受體激動(dòng)劑Neu-p11改善T2DM大鼠的胰島素抵抗作用研究
- 丹酚酸B通過抑制NLRP3炎癥小體priming階段減輕缺氧誘導(dǎo)大鼠心肌細(xì)胞損傷
- miR-142-3p靶向HMGB1逆轉(zhuǎn)乳腺癌細(xì)胞MCF-7對(duì)阿霉素的耐藥性
- 依托咪酯影響感覺刺激誘發(fā)小腦皮層分子層場(chǎng)電位變化的機(jī)制