煤焦瀝青煙提取物中多環芳烴含量的GC-MS法測定
2020-02-27 11:11:10萬珍珍吳擁軍李文杰屈凌波
鄭州大學學報(醫學版) 2020年1期
萬珍珍,李 亮,王 佳,劉 潔,于 斐,吳擁軍,李文杰,屈凌波
1)鄭州大學公共衛生學院營養與食品衛生學教研室 鄭州450001 2)鄭州大學化學與分子工程學院 鄭州450001
煤焦瀝青是經過蒸餾把煤焦油中的輕質組分提取后剩余的殘渣,含有多種多環芳烴[1-2]。多環芳烴指的是分子中含有兩個或者兩個以上苯環的碳氫化合物[3],產生于有機物不完全燃燒或熱解過程。美國環保署和歐盟已將其確定為優先控制毒物[4-7],因此多環芳烴的毒性作用及檢測方法研究顯得尤為重要。多環芳烴的分析方法主要有熒光光譜法、紫外分光光度法、高效液相色譜法、氣相色譜法和酶聯免疫吸附法等[8-11],其中氣相色譜-質譜聯用技術(GC-MS法)可以同時進行準確定性和定量,非常適合復雜組分中多環芳烴的分析。本實驗利用GC-MS技術建立了一種檢測煤焦瀝青煙中多環芳烴含量的方法,為優化煤焦瀝青的應用和研究其致病機制提供一定的方法學支持。
1 材料與方法
1.1實驗儀器與試劑7890B-5977A GC-MS儀(美國Agilent公司);BT125D電子天平[賽多利斯科學儀器(北京)有限公司];10~100、100~1 000 μL移液器(德國Eppendorf公司)。二氯甲烷(色譜純,天津市福晨化學試劑廠);12種多環芳烴標準品見表1,均購自美國Sigma公司,4 ℃密封避光保存。
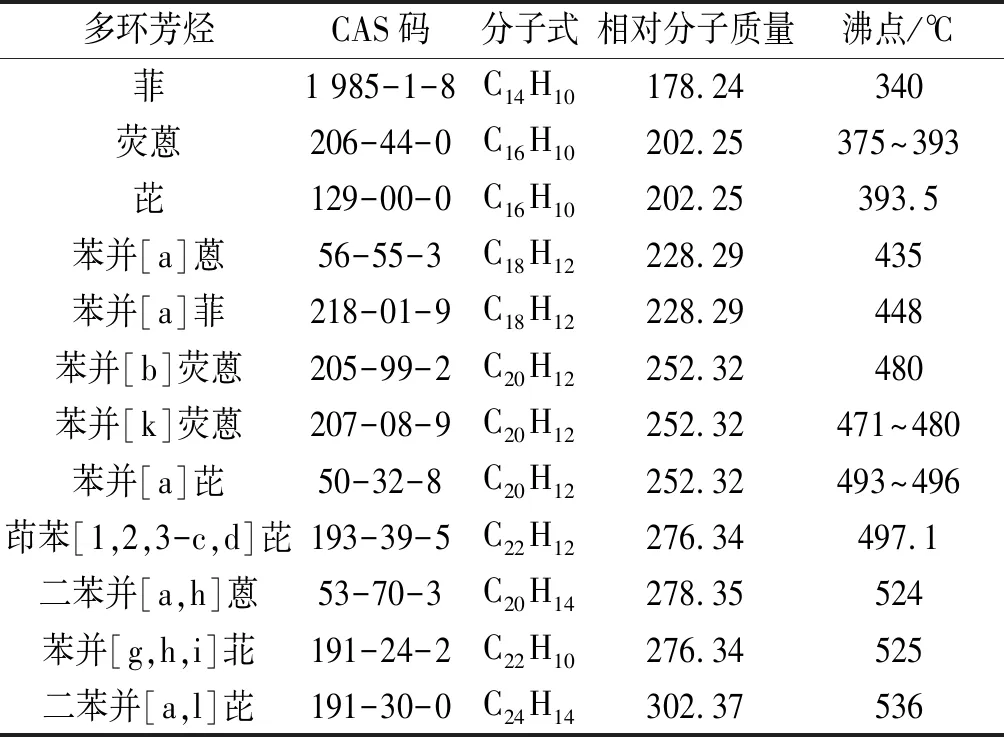
表1 12種多環芳烴標準品的性質
1.2實驗條件
1.2.1 色譜條件 色譜柱為DB-5MS石英毛細管柱(30.0 m×0.25 mm×0.25 μm);進樣口溫度為290 ℃;載氣為高純氦氣;流速為1.0 mL/min;進樣方式為不分流進樣;進樣量為1 μL;程序升溫為初溫120 ℃保持1 min,以10 ℃/min升至200 ℃保持2 min,以5 ℃/min升至300 ℃保持8 min。
1.2.2 質譜條件 離子源為EI;離子源溫度為230 ℃;傳輸線溫度為280 ℃;四極桿溫度為150 ℃;電子轟擊能量為70 eV;溶劑延遲為9 min。
1.3實驗方法
1.3.1 溶液的配制 用天平分別稱取菲、熒蒽、芘、苯并[a]蒽、苯并[a]菲、苯并[b]熒蒽、苯并[k]熒蒽、苯并[a]芘、苯并[g,h,i]苝標準品各1.00 mg,茚苯[1,2,3-c,d]芘、二苯并[a,h]蒽標準品各1.10 mg,二苯并[a,l]芘標準品1.30 mg,置于12個樣品瓶,用1 mL二氯甲烷溶解,制成12種標準儲備液,4 ℃避光保存備用。分別移取每種標準儲備液10 μL于樣品瓶中,加入990 μL二氯甲烷并混勻,配制成100、110 或130 μg/L的單樣品標準溶液,于4 ℃避光條件下保存備用。分別移取12種單樣品標準溶液50 μL于樣品瓶中,再加入二氯甲烷400 μL,配制成混合標準溶液①(簡稱混標①),再依次逐級稀釋,配制成濃度梯度溶液,見表2。
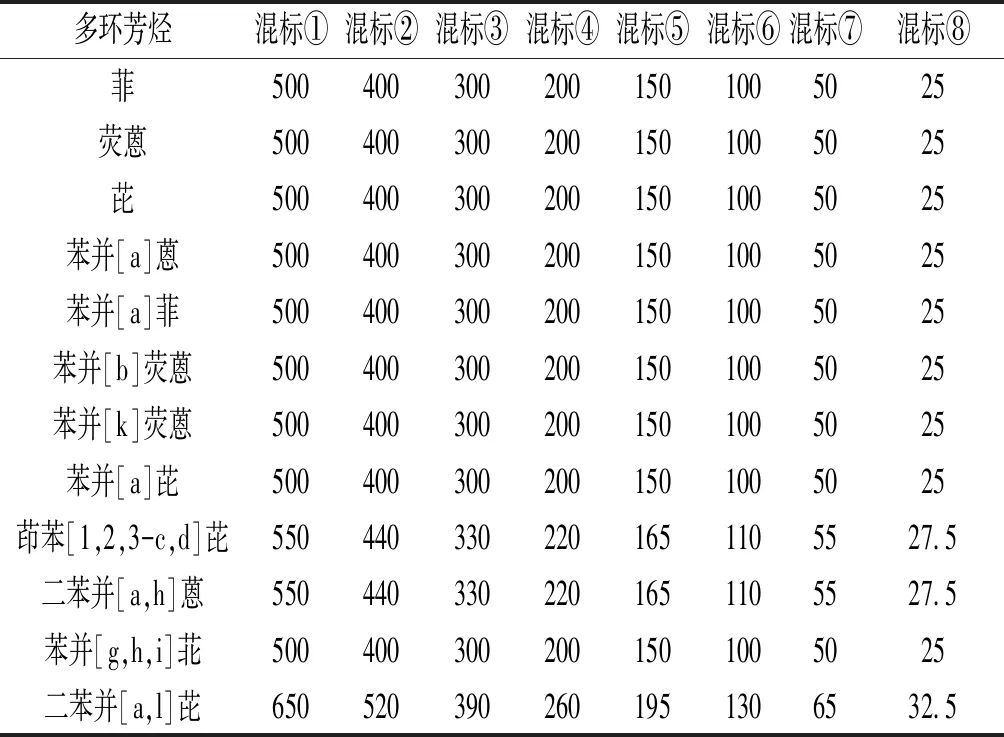
表2 混合標準溶液的濃度 μg/L
樣品溶液的配制:將煤焦瀝青加熱400 ℃,并將采樣后的濾膜剪碎, 置于具塞三角瓶中,加入約50 mL乙酸乙酯,超聲振動40 min,砂芯漏斗過濾, 經過提取后制成粗提液, 置于45 ℃烘干箱中揮發至干后用DMSO溶解得到10 g/L煤焦瀝青煙提取物。將制備好的10 g/L煤焦瀝青煙提取物作為原樣,通過將原樣稀釋不同倍數(25、50、100、125、200、250、500、1 000、2 000、2 500、4 000、5 000)分別配制12個樣品溶液。
1.3.2 色譜、質譜條件的優化 將12種標準儲備液稀釋100倍后依次進樣,采用全掃描模式(SCAN法)來優化色譜、質譜條件,得到峰形尖銳、分離度好的全掃描模式色譜圖和12種多環芳烴的質譜圖后確定12種多環芳烴的定性及定量離子質荷比。
1.4方法學檢驗
1.4.1 標準曲線的建立 采用外標法進行定量分析。在優化色譜、質譜條件下測定8種混合標準溶液,將混標溶液的測定結果導入MassHunter定量分析軟件,以各個組分的峰面積為Y、質量濃度為X進行線性回歸并做圖,得到12種多環芳烴的線性方程和決定系數R2。
1.4.2 檢出限的測定 采用信噪比法確定12種多環芳烴的檢出限。
1.4.3 精密度的測定 在優化條件下,采用同一條件對混標④進行6次平行測定,記錄12種多環芳烴的峰面積,計算RSD。
1.4.4 加標回收率的計算 采用1.2條件,在樣品溶液中分別加入低、中、高3個加標水平的多環芳烴進行加標回收實驗。菲、熒蒽、芘、苯并[a]蒽、苯并[a]芘、苯并[a]菲、苯并[b]熒蒽、苯并[k]熒蒽、苯并[g,h,i]苝分別加入50、100、150 μg/L;茚苯[1,2,3-c,d]芘、二苯并[a,h]蒽分別加入55、110、165 μg/L;二苯并[a,l]芘加入65、130、195 μg/L。每個水平平行測定3次取其平均值,計算加標回收率。
1.5樣品分析采用SIM模式在優化條件下對樣品溶液進行測定。依據定性、定量離子比例和保留時間進行定性分析,利用峰面積進行定量分析,每個樣品測定3次取平均值。將樣品溶液的檢測數據和混標溶液的數據導入MassHunter定量分析軟件,計算樣品溶液中12種多環芳烴的濃度。
2 結果
2.1色譜、質譜條件的優化在優化條件下,多環芳烴混標溶液中12種組分均得到了良好的分離,峰形尖銳,分析時間適中。確定的定性、定量離子質荷比及保留時間見表3。
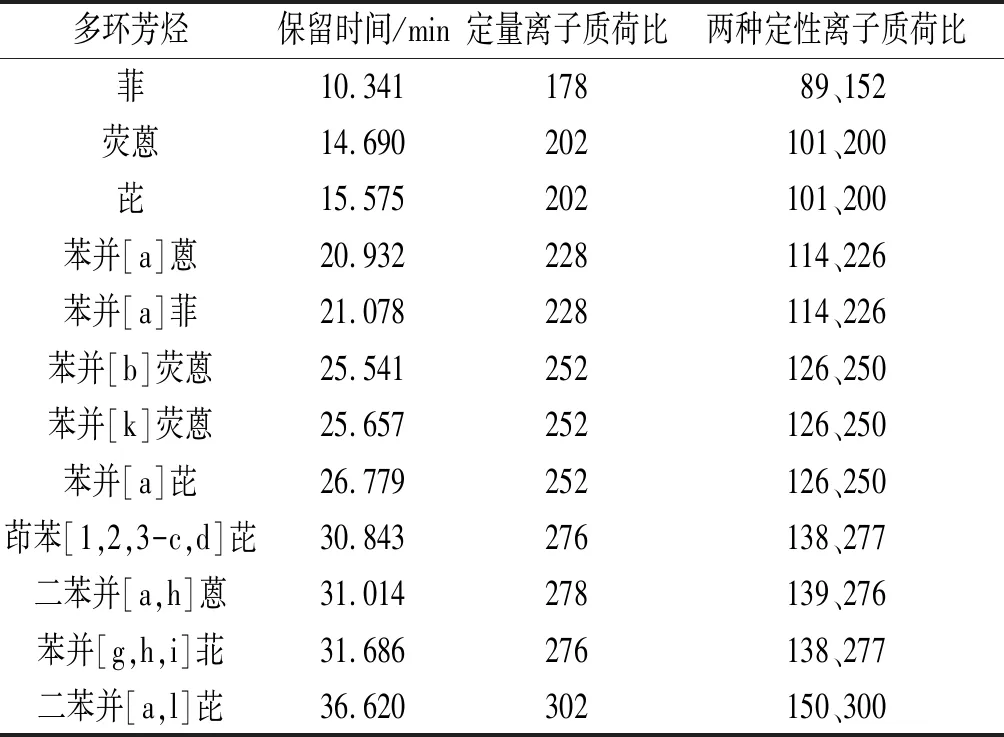
表3 12種多環芳烴的定量及定性離子
2.2方法學評價
2.2.1 標準曲線的建立 在優化條件下測定結果顯示,在一定濃度范圍內12種多環芳烴的回歸方程線性良好,見表4。
2.2.2 方法的檢出限 結果見表5。
2.2.3 精密度 結果見表5。從表5可以看出,方法的精密度較好,RSD均低于0.05%。
2.2.4 加標回收率 結果見表6。
2.3樣品分析通過本實驗方法測得煤焦瀝青煙提取物中菲、熒蒽、芘、苯并[a]蒽、苯并[a]菲、苯并[b]熒蒽、苯并[k]熒蒽、苯并[a]芘、茚苯[1,2,3-c,d]芘、二苯并[a,h]蒽、苯并[g,h,i]苝、二苯并[a,l]芘的含量分別為18.26、44.42、43.59、47.39、55.28、44.69、21.34、29.44、11.98、6.55、10.36、1.55 g/kg,占煤焦瀝青煙提取物總量的33.48%。

表4 12種多環芳烴的標準曲線
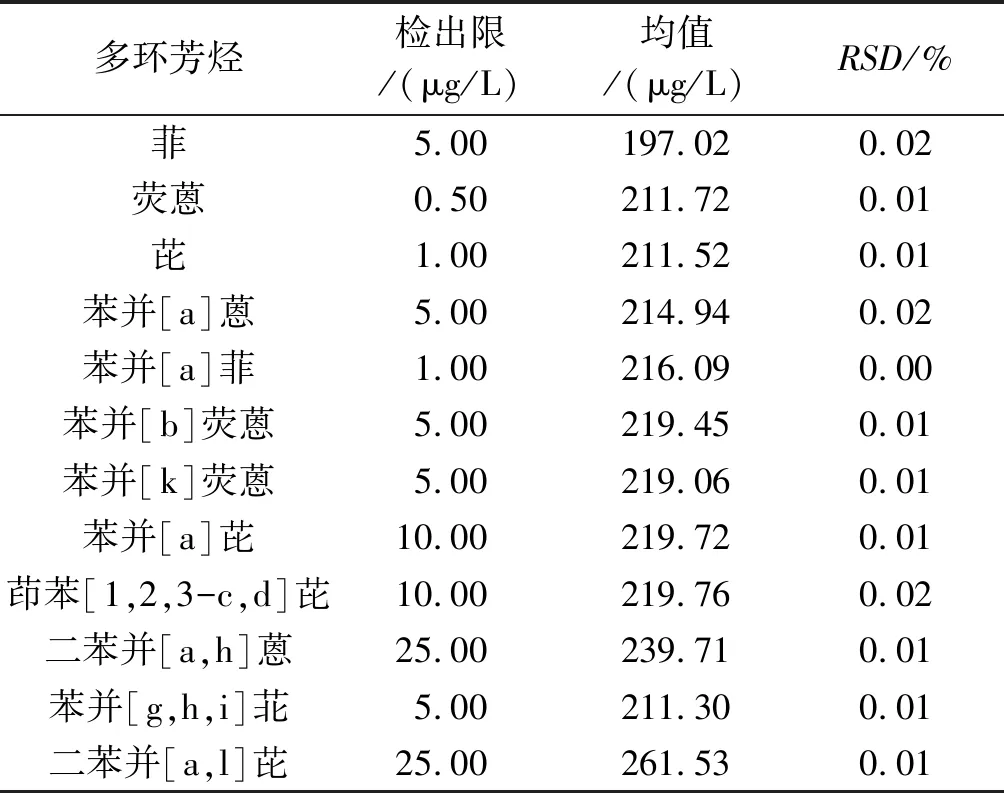
表5 12種多環芳烴的檢出限、均值和RSD
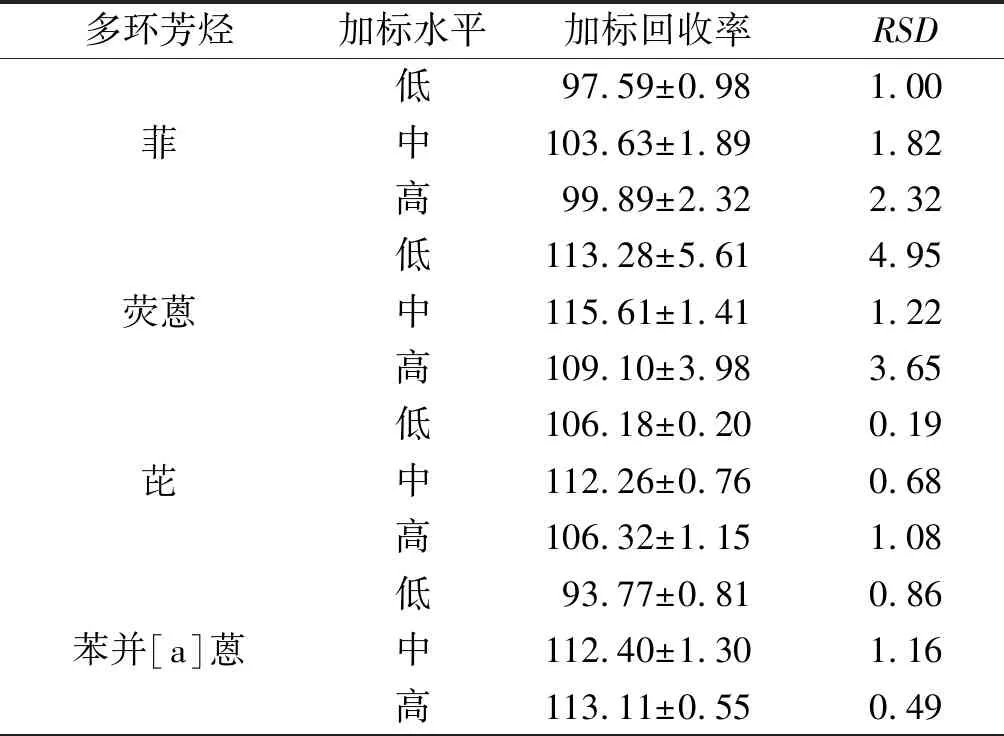
表6 12種多環芳烴的加標回收率(n=3) %
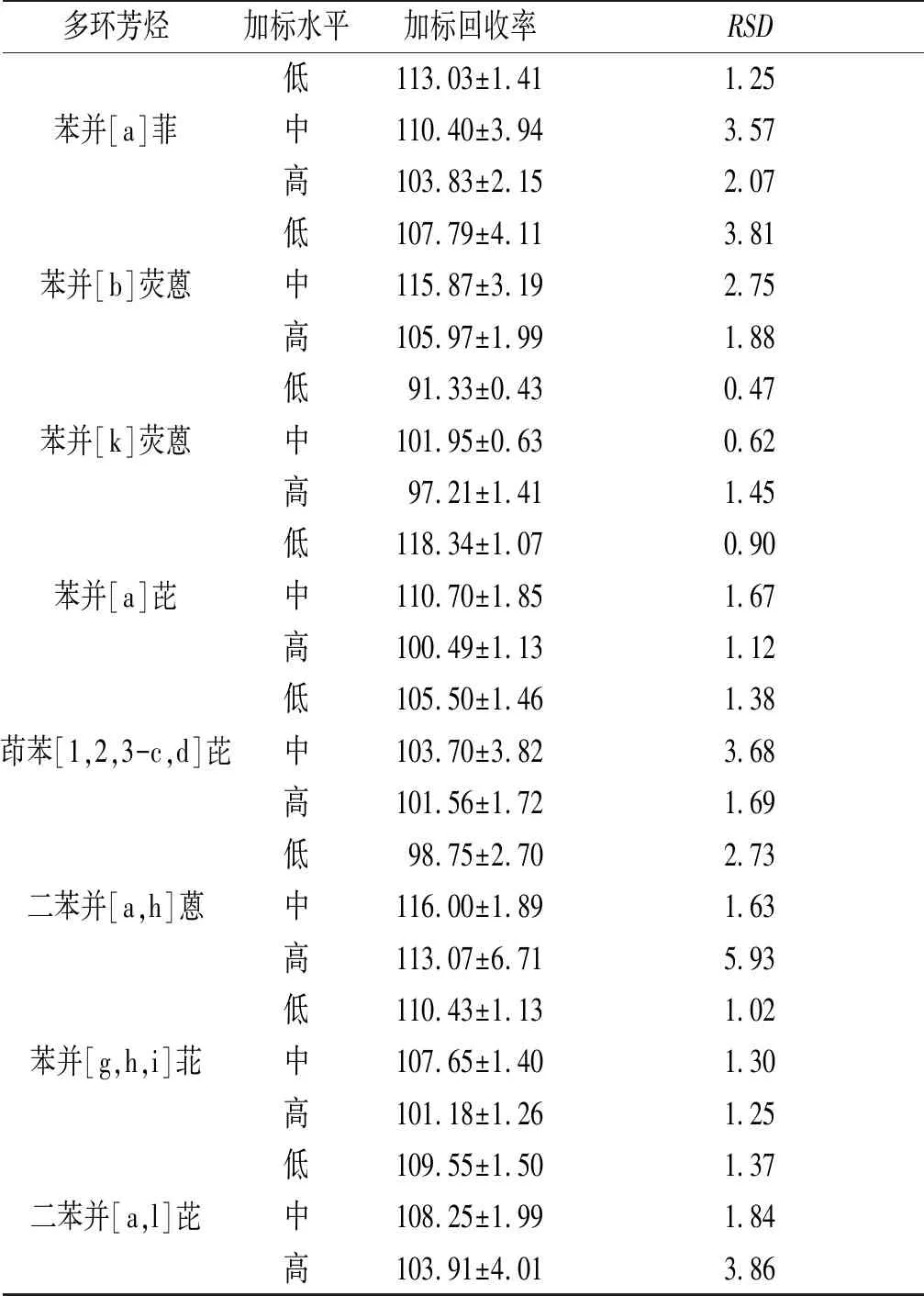
續表6
3 討論
本實驗采用GC-MS技術建立了快速、高效測定煤焦瀝青煙提取物中12種多環芳烴含量的方法。12種待測多環芳烴的沸點較高,沸程較寬,所以采用程序升溫的辦法。加快程序升溫速度可使各組分的保留時間前移,既節省實驗時間,又提高了工作效率。將進樣口溫度升至290 ℃可以有效降低進樣口的污染,還可提高部分高沸點組分的響應值。待測組分理化性質不同,氣化速度不同,在具體的分流比條件下各組分的實際分流比有差異,這就導致進入色譜柱的組分與樣品組分的比例不一致,使得檢測結果的定量分析不準確,而不分流進樣則具有更高的靈敏度[8]。
在優化條件下測得12種多環芳烴標準曲線的決定系數≥0.995 1;線性范圍為25~650 μg/L,檢出限最低可達0.5 μg/L;在精密度實驗中測得RSD均低于0.05%。說明該方法準確度高,靈敏度較好,回收率高,適用于實際樣品中多環芳烴的測定。
基于本實驗建立的方法測得煤焦瀝青煙提取物樣品中12種多環芳烴占煤焦瀝青煙提取物總量的33.48%。由此可見,煤焦瀝青煙提取物中多環芳烴的含量較高,這也是焦爐工人癌癥、多種慢性疾病及心血管疾病的發病率高于正常人群的主要原因[7]。因此,工廠在使用煤焦瀝青過程中應優化加工工藝,同時采取有效防護措施降低其對工人的危害。
