基于動態流變學的明膠/變性淀粉共混體系溶膠-凝膠相互轉變動力學解析
2020-03-01 21:27:26鄒秀容朱建華劉日斌
食品科學 2020年3期
關鍵詞:體系
鄒秀容,朱建華*,劉日斌,單 斌
(韶關學院英東食品科學與工程學院,廣東 韶關 512005)
食品工業所囊括的各種產品中凝膠態食品占據重要的份額,食品凝膠通常是由生物高分子(蛋白、多糖)為結構材料形成的物理凝膠,其主要通過改變溫度或調整離子強度、pH值等方法使生物高分子溶液體系完成溶膠-凝膠轉變而獲得。生物高分子體系溶液-凝膠相互轉變過程非常復雜,受到高分子組分類別、濃度、離子強度、pH值以及共溶質類型和濃度的影響[1],部分食品物理凝膠加熱時發生凝膠-溶膠相轉變,由凝膠態可逆回歸到溶膠態。
明膠軟糖、瓊脂軟糖及卡拉膠軟糖等凝膠糖果產品為典型的熱可逆物理凝膠,此類凝膠糖果產品生產制備過程常以生物高分子(蛋白、多糖)為主要結構材料成分,在共溶質(蔗糖、葡萄糖漿等)場中通過溶膠-凝膠成型制備[2]。共溶質的分子質量雖遠小于高分子組分,但可改變高分子組分在溶液中的分子折疊和纏結,并改變體系流變性質,進而影響到產品質構、風味釋放等性質[3-4]。明膠軟糖通常具備良好的彈性和耐咀嚼性等質構特性,廣受消費者青睞。明膠軟糖的主要成分明膠是由膠原質通過加熱酸法或堿法水解獲得,膠原質分子的天然構象主要由3 條多肽鏈形成的三螺旋結構構成并通過氫鍵穩定,多肽鏈上每3 個殘基的重復序列為甘氨酸-脯氨酸-羥基脯氨酸(Gly-Pro-Hyp),水解之后獲得的明膠分子在水中呈直鏈高分子狀,具有無規卷曲構象[5]。
從材料學視角看,明膠軟糖為一類天然高分子物理凝膠,其溶膠-凝膠制備過程原理主要涉及明膠分子發生線團-螺旋轉變。明膠軟糖制備過程單一使用明膠作為食品高分子基材時,其最終制品質構特性常存在彈性過大、咀嚼性及成本高等劣勢,難以滿足消費者和生產者需求,為此,食品工業生產過程通常采用明膠與變性淀粉復配來改善產品口感及降低成本[6]。復配過程實際上借鑒了材料科學研究領域的聚合物共混改性技術,以發揮明膠及變性淀粉各自的物性優點。但此類產品貨架期容易出現析水、發烊等質量劣化問題,此類質量問題的突出特征為結構變形、崩解及液化而黏連外包裝等,主要由制品發生凝膠-溶膠相轉變導致結構解離引起。因此開展明膠/變性淀粉復合凝膠糖果溶膠-凝膠及凝膠-溶膠相轉變過程特性研究具有重要的學術和應用價值。
小振幅動態流變測試因對受試材料自身結構不會造成破壞,且試樣可呈現線性黏彈性質,對其形態結構響應性非常敏感,常被用于表征高分子材料的結構形態。近年來對明膠-多糖共混體系相關性質的研究引起關注[7],但迄今為止鮮見文獻報道應用動態流變學研究共溶質場中明膠/多糖共混體系相轉變動力學。本實驗研究葡萄糖漿共溶質場中明膠/變性淀粉雙組分模型共混體系溶膠-凝膠相互轉變過程流變性質變化趨勢,進而分析相轉變動力學,以期為明膠軟糖類凝膠制品工藝優化及質量控制提供應用理論指導。
1 材料與方法
1.1 材料與試劑
B型明膠(食品級、200 g凍力、pI 4.4~4.7、水分質量分數10.5%、灰分質量分數1.5%) 羅賽洛(廣東)明膠有限公司;DM-803酸法變性淀粉(modified starch,MS)(水分質量分數≤14%、灰分質量分數≤0.5%,80 目篩通過率≥95%) 東莞東美食品有限公司;葡萄糖漿(glucose syrup,GS)(淀粉水解度42%) 廣州雙橋股份有限公司。
1.2 儀器與設備
SRT-202滾軸混合器 其林貝爾儀器制造有限公司;SHA-C恒溫水浴振蕩器 常州國華儀器有限公司;MCR92旋轉流變儀 奧地利Anton Paar公司。
1.3 方法
1.3.1 明膠-變性淀粉共混溶液體系制備
12%(質量分數,下同)明膠儲備液在80 ℃水浴中恒溫30 min,以確保明膠顆粒全部水化溶脹分散。稱取適量變性淀粉分散于純水中,于90 ℃磁力攪拌水化5 min,配制均一的12%變性淀粉儲備液。將明膠儲備液和變性淀粉儲備液等體積混合于燒杯中,在80 ℃下混合均勻并保溫,獲得6%明膠/6%變性淀粉儲備液共混體系,隨后加入相應葡萄糖漿混勻,配制含0%(對照)、20%、40%、60%及80%葡萄糖漿共溶質的明膠/變性淀粉共混體系。
1.3.2 動態流變學參數測定
含有不同質量分數葡萄糖漿的明膠-變性淀粉共混溶液于80 ℃恒溫30 min后轉移至保溫桶中進行保溫,然后將1 mL樣品快速轉移到預先加熱到75 ℃的流變儀平行板(上、下板直徑均為45 mm,狹縫寬度1 mm),除去過量樣品并在樣品外緣加一層輕質硅油防止樣品中的水分蒸發。保持在線性黏彈區范圍內的應變(γ)條件下(γ=0.5%)進行動態流變測試。
溶膠-凝膠轉變過程測試程序:首先于75 ℃以2 ℃/min降溫至5 ℃,然后在5 ℃恒溫20 min,此過程角頻率(ω)為6.28 rad/s,記錄隨時間(t)變化的彈性模量(G’)、損耗模量(G”)及損耗系數(tanδ);恒溫末進行頻率掃描,分析共混體系力學響應性質,頻率掃描范圍為0.1~100 rad/s,記錄G’、G”及復數黏度(η*)隨ω變化的情況。
凝膠-溶膠轉變過程測試程序:前置降溫恒溫程序同上述溶膠-凝膠轉變測試,然后從5 ℃以2 ℃/min升溫至55 ℃,ω為6.28 rad/s,記錄隨t變化的G’、G”、tanδ;升溫末進行頻率掃描,頻率掃描范圍為0.1~100 rad/s,記錄隨ω變化的G’、G”及η*。
1.3.3 共混體系相轉變過程結構形成及解離速率的計算
非等溫或等溫過程特定時間階段共混體系凝膠或溶膠形成的相對快慢常用結構化平均形成速率(SDRa)、即時速率(v)進行表征[8-9]。SDRa可根據式(1)計算。

式中:G’ini及G’end分別為特定階段對應的起始、終止G′/Pa;tini及tend分別為對應階段起始、終止時間/s。
v可由式(2)計算。

式中:v為G’-t曲線求一階導數得到的結構化即時速率/(Pa/s),v>0時為凝膠化速率,以vg表示;v<0時為溶膠化速率,以vs表示。
SDRa及v分別可用于表征時間段及時間點的結構化相對快慢,但尚難以定量表征結構化速率改變的快慢,為此引入結構化加速度(α),按式(3)計算。

式中:α為對G’-t曲線求二階導數所得,記為即時加速度/(Pa/s2),加速度的正負性具有物理意義,其符號與v相同時表示結構化形成(或解離)處于加速階段;其符號與v相反時視為結構化形成(或解離)處于減速階段,用來輔助分析凝膠化或溶膠化加速過程特征。
1.3.4 溶膠-凝膠轉變及凝膠-溶膠轉變過程活化能的計算
降溫過程非等溫動力學模型常用于溶膠-凝膠轉變動力學分析[10]。此種方法主要基于經典的非等溫化學分解反應動力學方程[11](式(4))。
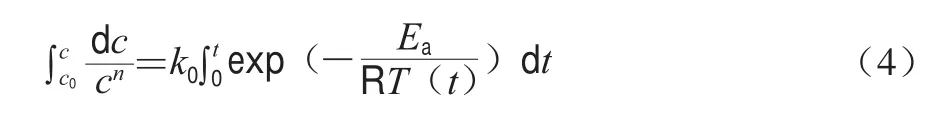
式中:c為反應物濃度/(mol/L);c0為反應物初始濃度/(mol/L);t為反應時間/s;k0為指前因子;Ea為反應活化能/(J/mol);T為t時刻的絕對溫度/K;R為氣體常數(8.314 J/(molgK)),此表達式依賴于變溫速率。變溫速率恒定的前提下,基于實驗實證和回歸分析推導獲得式(5),其中Ea和k0可由Arrhenius關系圖中計算,即通過對1/T作圖確定。
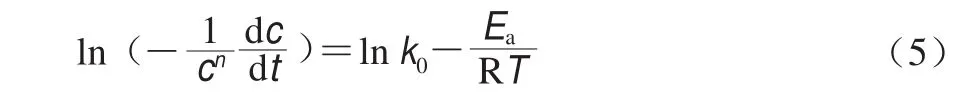
非等溫條件下的經典化學反應動力學模型與流變測試黏彈性模量測試可借鑒橡膠彈性理論進行關聯轉換。具體就溶膠-凝膠相互轉變體系而言,因G’通常與高分子交聯結構數量或濃度成比例,因此濃度改變效應可通過同步改變的G’來研究,對方程(5),G’與c的改變具有等效性,且c和其變化值(dc)可進一步用G’及其變化值(dG’)予以替代[12],并得到直接用流變學參數表征相轉變過程的動力學方程(式(6))。
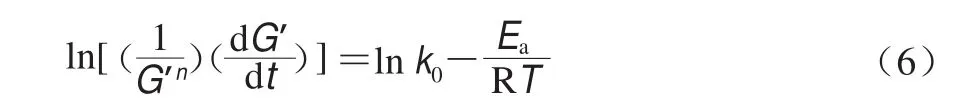
與公式(5)相比,溶膠-凝膠轉變過程G′隨時間延長呈增長趨勢,此公式中負號轉換為正號是因為凝膠形成過程G’增量為正,這與反應物濃度增量為負相反。生物大分子溶膠-凝膠過程通常取n=2[13-14]。物理凝膠的凝膠-溶膠轉變過程因G’隨時間延長呈遞減趨勢,此公式中仍保留負號是因為凝膠形成過程G’增量為負,與反應物濃度增量為負一致。
1.4 數據統計與分析
實驗數據采用Origin 9.0軟件作圖,流變曲線及Ea求解擬合過程均采用最小二乘法。
2 結果與分析
2.1 葡萄糖漿共溶質質量分數對明膠/變性淀粉共混體系溶膠-凝膠轉變過程流變性質的影響
2.1.1 溶膠-凝膠轉變過程G’、G”隨t的變化分析結果
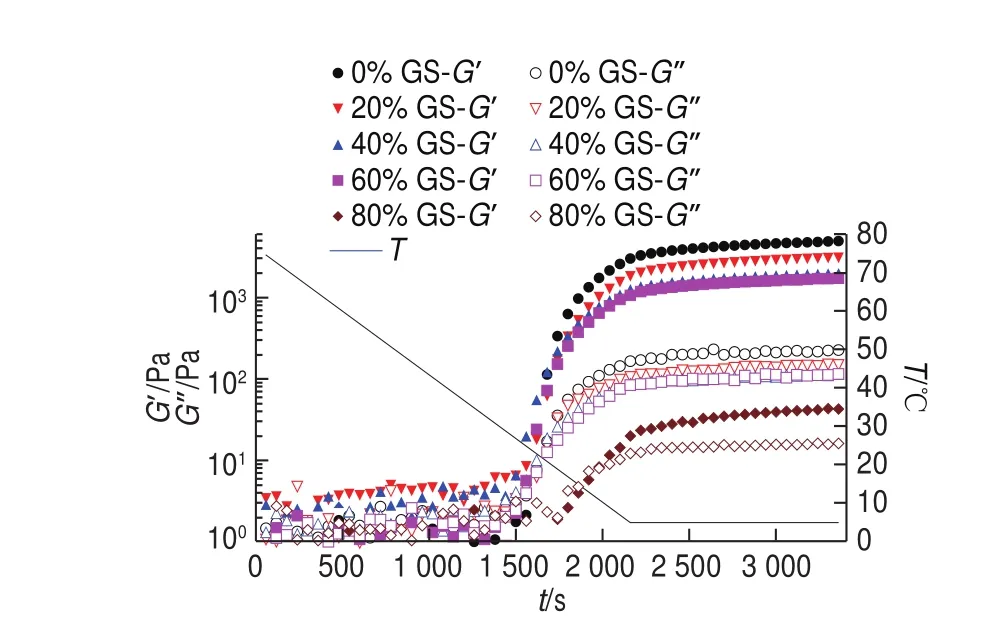
圖 1 明膠/變性淀粉共混體系溶膠-凝膠轉變過程G’、G”隨t變化曲線Fig. 1 G’ and G” versus t cures of GL/MS blends during sol-gel transition
由圖1可知,不同質量分數葡萄糖漿共溶質場中明膠-變性淀粉共混體系降溫階段G’、G”-t曲線均為類“S”型。0~1 500 s階段(約75~30 ℃)G’、G”相對較小,均低于10 Pa,共混體系仍然為可流動的濃溶液狀態,明膠及變性淀粉分子主要處于無規卷曲狀態。隨溫度下降,到特定溫度點時,G’、G”從低模量基準線躍增。第二階段隨溫度降低G’、G”均呈增加趨勢,且差幅越來越小,G’=G”的溫度點被定義為凝膠點(Tg)[15]。此溫度點溶膠體系首次形成三維網絡結構,主要因為共混體系中形成的三螺旋交聯區域覆蓋到整個溶液共混體系空間,共混體系不能再流動。隨溫度進一步降低直至接近降溫程序末端(5 ℃),G’、G”進一步增加,且G’與G”增幅差逐漸加大,主要因為非凝膠區域逐漸有更多的明膠高分子形成三螺旋并加入和充密到既有三維網絡空間所致[16]。結合表1可知,與未添加葡萄糖漿組相比,20%葡萄糖漿共溶質存在時共混體系G’1800s、G’降溫末、G’恒溫末由633.25、3 044.1 Pa及5 139.5 Pa分別降低為343.17、1 898.4 Pa及3 168.6 Pa,降低幅度分別達45.80%、37.64%及61.65%,且SDRa降溫由1.41 Pa/s降低至0.88 Pa/s。隨葡萄糖漿質量分數增加至40%~60%,G’1800s、G’降溫末及G’恒溫末進一步降低。80%葡萄糖漿共溶質存在時,與添加60%葡萄糖漿樣品相比,G’降溫末由1 074.3 Pa驟降至19.9 Pa,降低近兩個數量級,表明添加80%葡萄糖漿共混體系形成的凝膠三維網絡結構非常弱。
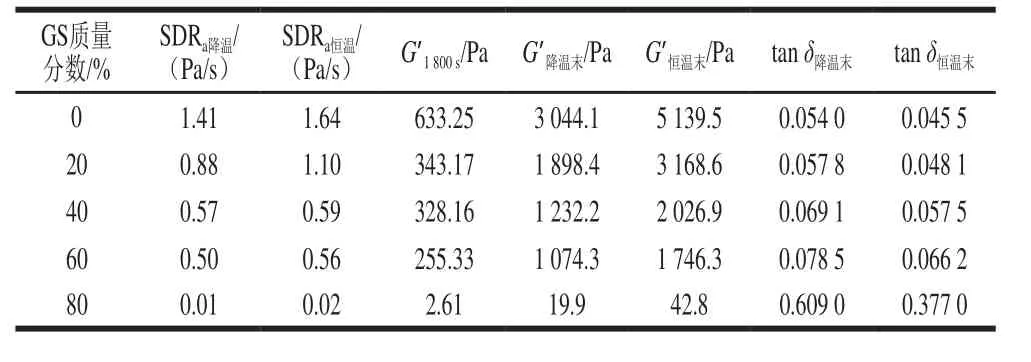
表 1 明膠/變性淀粉共混體系溶膠-凝膠轉變過程流變學特征參數Table 1 Rheological characteristic parameters of GL/MS blends during sol-gel transition
2.1.2 溶膠-凝膠轉變過程特征區域分析結果
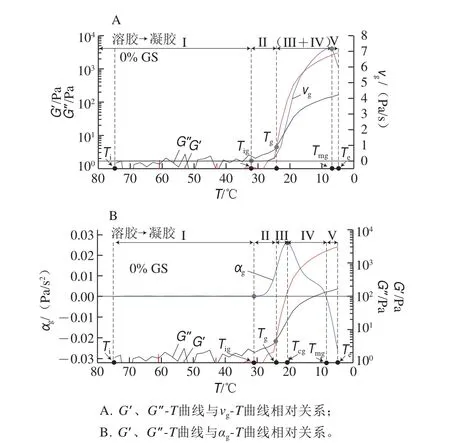
圖 2 明膠/變性淀粉共混體系溶膠-凝膠轉變特征溫度解析圖Fig. 2 Schematic illustration of the characteristic temperatures of GL/MS blends during sol-gel transition
G’、G”-T測定結果可較為清晰地定性分析明膠/變性淀粉共混體系溶膠-凝膠轉變結構形成過程,將溶膠-凝膠起始階段G’-T曲線分為誘導期、快速指數增長期及對數緩慢增長期,可以確定Tg點,但尚不能定量描述結構改變過程即時結構形成速率及其他關鍵溫度點。為此對G’-T曲線求一階導數和二階導數,并轉化得到相轉變結構化速率曲線(v-T)和加速度曲線(α-T)。圖2以未添加葡聚糖共混體系降溫過程的vg-T、αg-T為例,對溶膠-凝膠轉變過程進行分析。曲線vg-T或αg-T外推與vg=0或αg=0水平線(零線)相交的溫度點定義為凝膠化起始點溫度(Tig),此溫度點確定方式示意圖參照圖3A中內置圖;G’-T與G″-T曲線交點對應溫度為凝膠形成點溫度(Tg);vg-T曲線上vgmax對應溫度定義為Tmg;αg-T曲線上αgmax對應溫度定義為Tcg,此溫度點不能直接從vg-T曲線上得出拐點對應溫度,僅能從αg-T曲線上解析出。圖2解析結果表明降溫階段除去降溫程序起始溫度點(Ti)和程序終點溫度(Te),存在上述4 個特征溫度Tig、Tg、Tcg及Tmg,溶膠-凝膠相轉變過程5 個階段的溫度順序為Tig>Tg>Tcg>Tmg。
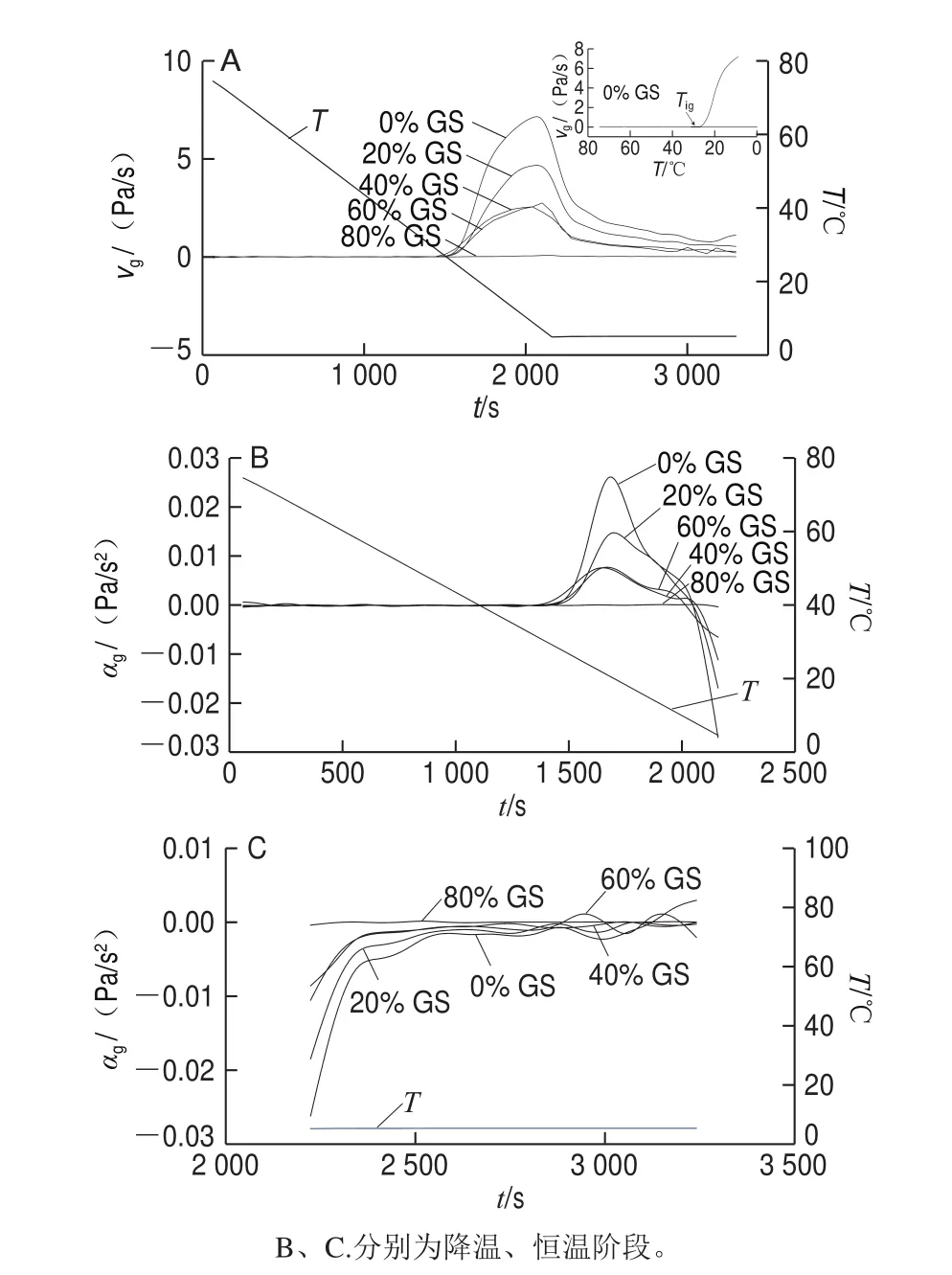
圖 3 明膠/變性淀粉共混體系凝膠化速率(A)及加速度(B、C)隨t變化曲線Fig. 3 vg versus t (A) and αg versus t (B, C) cures of GL/MS blends

表 2 明膠/變性淀粉共混體系溶膠-凝膠轉變過程特征溫度及凝膠結構化參數Table 2 Characteristic temperatures and structure formation parameters of GL/MS blends during sol-gel transition
圖3為降溫恒溫過程明膠/變性淀粉共混體系vg-t或αg-t曲線,表2為該過程結合圖3解析得出的特征參數。與對照組相比,添加20%葡萄糖漿樣品Tig由27.50 ℃提高至31.28 ℃,可能的原因是葡萄糖漿存在時凝膠結構化起始溫度得到提高。隨葡萄糖漿質量分數進一步增加至40%及60%,Tig進一步被提高,0%~60%葡萄糖漿質量分數范圍Tg、Tcg、Tmg變化趨勢與Tig相同,隨共溶質質量分數增加,各特征溫度均被提高,表明此質量分數范圍葡萄糖漿中低分子糖類各成分存在大量的羥基與水分子形成較強氫鍵作用,打破了明膠高分子與水分子的相互作用,促進高分子由無規卷曲態向螺旋過渡態轉變,并推動三螺旋聚集體形成,最終促成結構形成各特征溫度提高。但80%葡萄糖漿存在時,與其他樣品相比,顯然共混體系凝膠結構形成被遲滯,Tig下降至26.10 ℃,Tg、Tcg、Tmg亦明顯降低,共混體系凝膠SDRa急速降低。出現凝膠結構發展起始溫度偏高但G’、SDRa、αg降低的可能原因主要是葡萄糖漿共溶質存在時明膠過渡態聚集物的長度減小,進而導致三螺旋聚集分子鏈縮短、三維網絡結構弱化所致。不同體系小分子共溶質對高分子凝膠強度的影響作用及原因存在明顯差異,Sharma等認為共溶質的存在阻斷瓊脂單股螺旋聚集,從而明顯降低螺旋交聯點的數量和密度,導致G’下降[17];Trombetta等通過跟蹤水分活度發現多糖分子外圍水化層水分子的減少限制了分子間螺旋的形成,并阻礙三維網絡結構的形成[10];Evageliou等發現質量分數低于40%的葡萄糖漿對κ-卡拉膠凝膠網絡結構可起到強化作用,主要因為其促進了卡拉膠分子無序-有序轉變,但高于此值時G’下降[11]。
2.1.3 恒溫階段溶膠-凝膠轉變流變性質分析結果
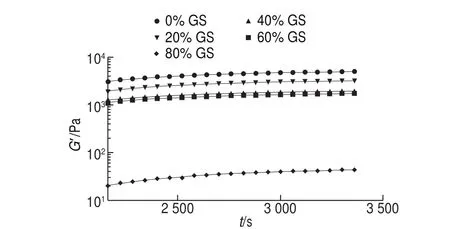
圖 4 恒溫階段明膠/變性淀粉共混體系G’隨t變化的擬合曲線Fig. 4 Fitted G’ versus t cures of GL/MS blends at constant temperature stage
圖4為共混體系在恒溫階段中G’隨t的變化曲線。結果表明雖然共混體系中含葡萄糖漿共溶質質量分數不同,但恒溫過程中G’隨t的變化規律非常相似。恒溫階段G’、G”增加進一步放緩,并趨近于平衡模量值。恒定溫度條件下,交聯區域的微區域調整重排及部分鏈段松弛,凝膠結構進入熟化和強化階段,Goudoulas等研究明膠-海藻酸鹽共混體系相轉變動力學過程發現了類似的現象[18]。據圖4中的曲線形狀,G’-t關系符合式(7)關系。

式中:G’為恒溫過程任意時刻的G’/Pa;G’∞為t=∞時的G’/Pa;m和k為大于零的常數,其中m反映凝膠結構達到G’∞時所需時間,而k為速率常數,反映G’隨t變化速率的大小。
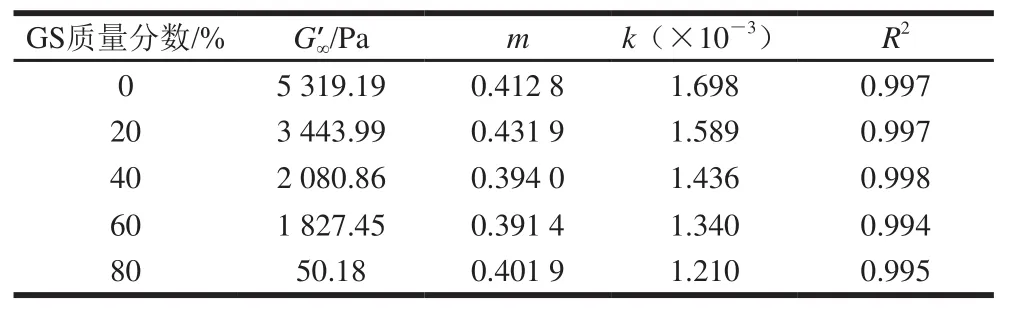
表 3 恒溫階段明膠/變性淀粉共混體系G’-t擬合曲線相關系數Table 3 Correlation coefficient of the G’-t fitted curves for GL/MS blends at constant temperature stage
用式(7)對圖4中各曲線進行擬合并分析,得到相關系數結果見表3,隨葡萄糖漿質量分數增加,G’∞及k逐漸明顯減小,與降溫過程速率變化的規律一致,說明葡萄糖漿質量分數越大,共混體系在恒溫過程形成的凝膠結構越弱,且凝膠速率也明顯降低。但是m隨葡萄糖漿質量分數增加變化較小,不同共溶質質量分數共混體系轉變至平衡凝膠結構的時間相當。等溫階段達到平衡結構的時間和凝膠強度無直接關聯,主要是因為共溶質的存在削弱了凝膠強度,同時凝膠結構化速率也被阻滯。
2.1.4 恒溫末共混體系頻率掃描力學性質分析結果
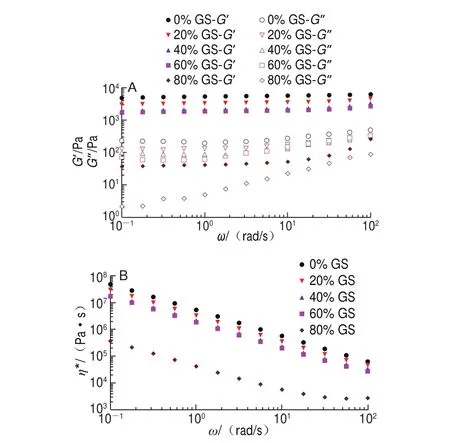
圖 5 明膠/變性淀粉共混體系恒溫程序末黏彈模量-ω(A)及η*-ω(B)曲線Fig. 5 G’ and G” versus ω (A) and η* versus ω (B) curves at the end of constant temperature stage
G’、G”及η*與ω的關系可用來反映受試材料的結構類型。恒溫末頻率掃描圖(圖5A)顯示除添加80%葡萄糖漿共混體系樣品在高頻范圍對頻率依賴性較高外,其余樣品全頻率掃描范圍G’、G”對ω的依賴均很小,且隨葡萄糖漿質量分數增加G’逐漸降低。圖5B顯示對照組的η*均高于添加共溶質組,η*隨ω的增加而減小,且整個頻率范圍0%~60%葡萄糖漿共混體系樣品η*-ω雙對數擬合直線斜率約為-1,表明均可形成較好的凝膠結構[19],但80%葡萄糖漿樣品的η*在高頻范圍出現平臺區,顯示類似剪切變稀性質,表明凝膠結構非常弱。
2.2 葡萄糖漿共溶質質量分數對明膠/變性淀粉共混體系凝膠-溶膠轉變過程流變性質的影響
2.2.1 凝膠-溶膠轉變過程G’、G”隨t的變化分析結果
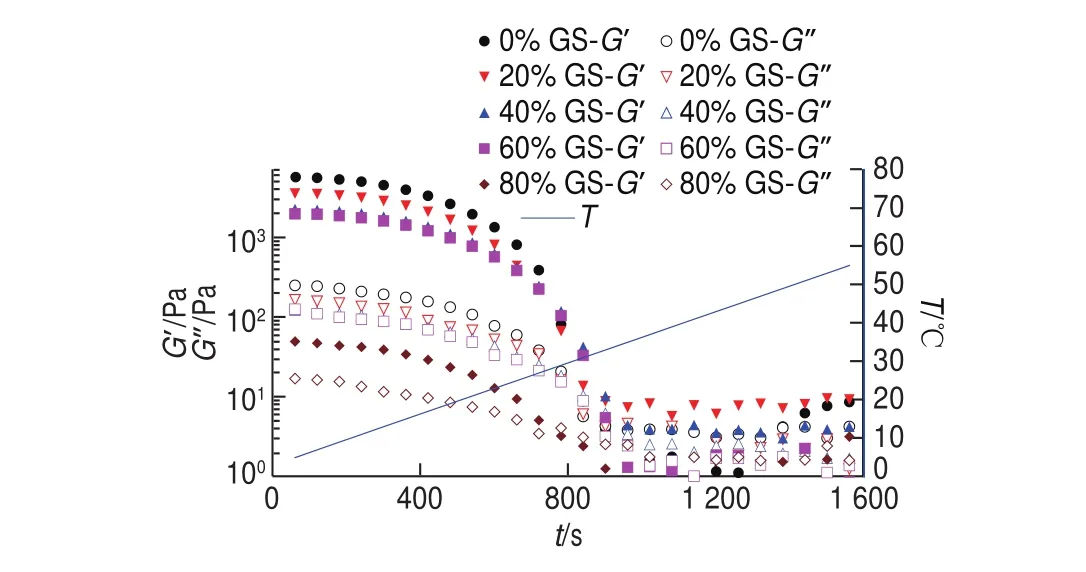
圖 6 明膠/變性淀粉共混體系凝膠-溶膠轉變過程G’、G”隨t變化曲線Fig. 6 G’ and G” versus t cures of GL/MS blends during gel-sol transition
圖6顯示,明膠/變性淀粉共混體系于5~55 ℃升溫區間凝膠-溶膠相轉變過程G’、G”隨t變化,升溫初期(5~18.5 ℃)G’、G”及兩者差值開始呈減小趨勢。隨溫度增加至18.5~30 ℃,G’、G”及兩者差值進一步下降,但G′改變速率加大,直至G’=G”即為傳統定義的溶膠點臨界溫度(Ts),此溫度點維持臨界三維網絡結構,為體系可流動或形變的分界點。與溶膠-凝膠過程相比,添加相同葡萄糖漿質量分數共混體系的Ts均高于凝膠溫度Tg,研究人員曾在明膠等熱可逆凝膠體系中發現類似的溶膠點溫度滯后于凝膠點溫度的現象[20-21]。隨溫度進一步升高,G’、G”均降低且G’<G”,共混體系逐漸趨于溶膠化狀態,最終明膠三螺旋結構不存在于體系中,明膠變性淀粉共混體系進入濃溶液狀態,明膠分子的主要存在形式是無規卷曲。如表4所示,與對照組相比,添加20%葡萄糖漿的樣品,G’升溫始、G’t=300s、G’t=720s分別由5 651.9、4 489.9、386.5 Pa降低至3 643.2、2 934.8、225.8 Pa,平均溶膠化速率由3.77 Pa/s降低為2.43 Pa/s。80%葡萄糖漿共溶質存在時,樣品的tanδ升溫始明顯大于添加0%~60%葡萄糖漿樣品的對應值,表明80%葡萄糖漿樣品升溫過程中共混體系結構狀態明顯異于0%~60%樣品,三維網絡結構非常弱。
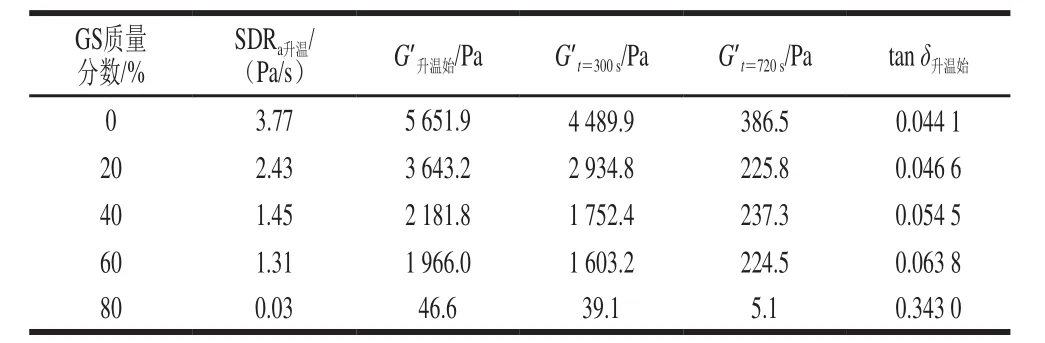
表 4 明膠/變性淀粉共混體系凝膠-溶膠轉變過程流變學特征參數Table 4 Rheological characteristic parameters of GL/MS blends during gel-sol transition
2.2.2 凝膠-溶膠轉變過程特征區域分析結果
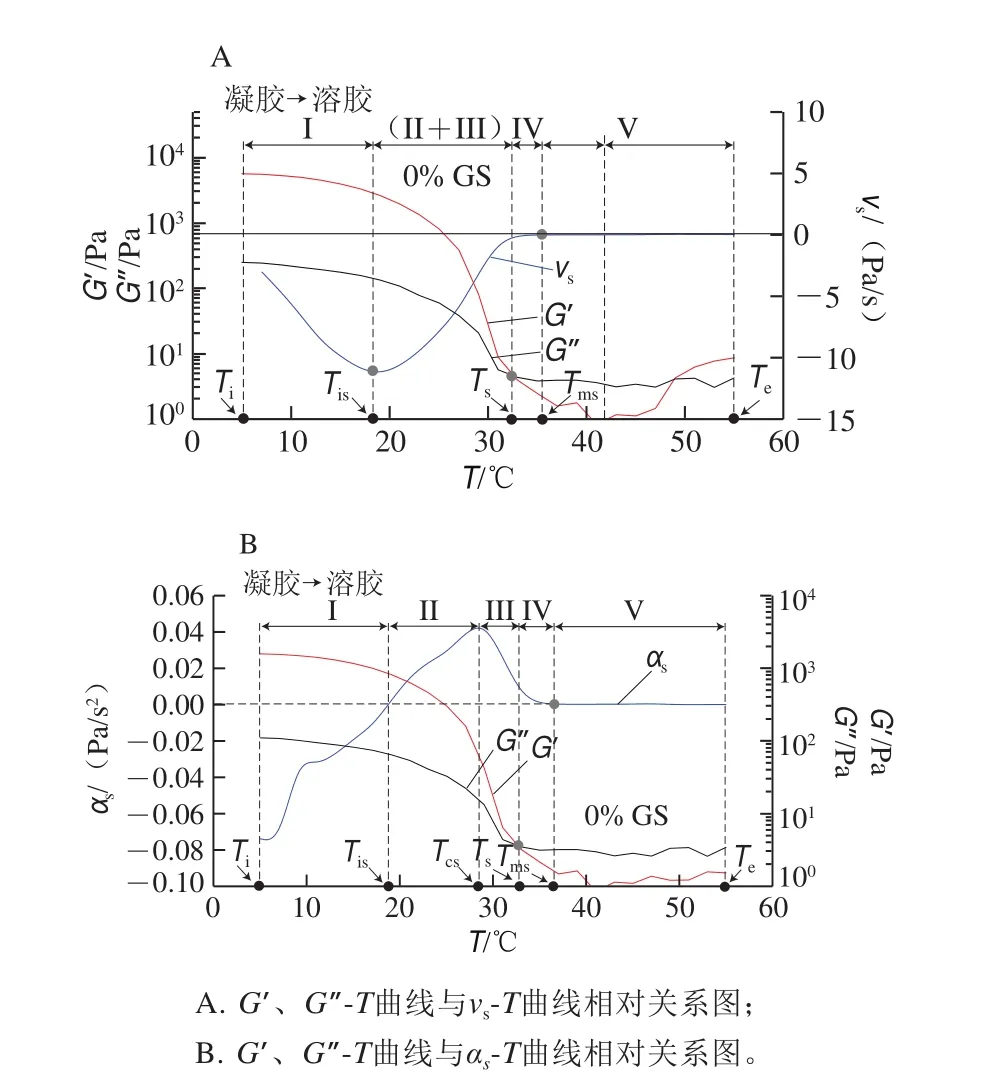
圖 7 明膠/變性淀粉共混體系凝膠-溶膠轉變特征溫度解析圖Fig. 7 Schematic illustration of the characteristic temperatures of GL/MS blends during gel-sol transition
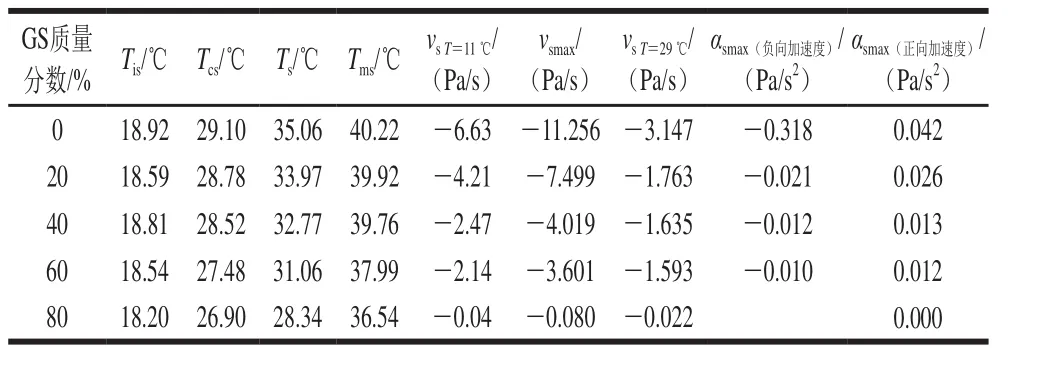
表 5 明膠/變性淀粉共混體系凝膠-溶膠轉變過程特征溫度及凝膠結構化參數Table 5 Characteristic temperatures and structure formation parameters of GL/MS blends during gel-sol transition
以未添加葡萄糖漿共溶質共混體系樣品為例,通過對凝膠-溶膠過程G’-T曲線進行一階和二階求導得到溶膠化速率和加速度曲線,并獲得另外4 個特征溫度參數(圖7):Tis為預溶膠起始關鍵溫度點,即溶膠化速率達最大值時對應溫度;Tcs為溶膠化加速度達最大值時對應溫度點;Ts為溶膠態形成溫度點,由G’-T、G”-T曲線交點確定;Tms為溶膠態完全轉變點溫度,由曲線vs-T或αs-T外推與vs=0或αs=0(零線)相交的溫度點。這4 個特征溫度值高低排序為Tis<Tcs<Ts<Tms,并將凝膠-溶膠化轉變過程分為5 段。含不同質量分數葡萄糖漿共混體系特征溫度見表5。
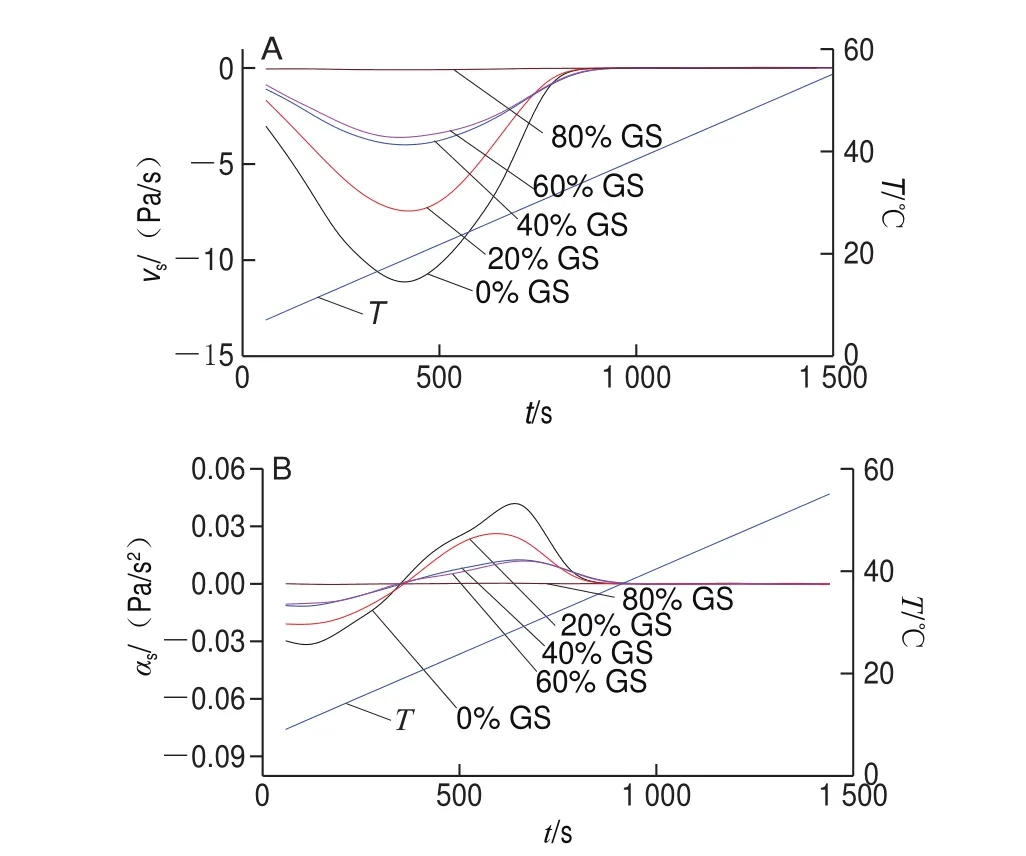
圖 8 明膠/變性淀粉共混體系溶膠化速率(A)及加速度(B)隨t變化曲線Fig. 8 vs versus t (A) and αs versus t (B) cures of GL/MS blends
圖8及表5結果表明,隨葡萄糖漿質量分數升高,溶膠化速率及溶膠化加速度均呈降低趨勢。葡萄糖漿共溶質對Tis影響甚微,但明顯降低了溶膠化速率,表明共溶質對預解離聯結區形成沒有影響,原因在于此過程僅涉及到構象熵調整,明膠分子鏈運動尚未開始。與對照組相比,添加20%葡萄糖漿后,明膠/變性淀粉共混體系的Ts由35.06 ℃降低至33.97 ℃,隨共溶質質量分數增加,Tcs、Ts、Tms均呈降低趨勢,其中對Ts影響最大,表明隨葡萄糖漿質量分數增加,溶膠化進程被提前且呈加劇趨勢,共溶質存在時利于凝膠結構發生解離。
2.2.3 升溫末共混體系頻率掃描力學性質分析
圖9顯示升溫末G’、G”及η*隨剪切速率增加呈現出兩個具有明顯差別的特征區域。當頻率小于10 rad/s時,對照組及添加20%葡萄糖漿樣品的流動性非常接近,但葡萄糖漿質量分數增加至40%、60%后,共混體系G’、G”對頻率依賴性逐漸增強,η*下降明顯,且提前到達平臺區;葡萄糖漿質量分數增加至80%后,η*重新上升,可能由葡萄糖漿質量分數進一步增加后導致共混體系的黏度值升高而引起。當頻率在10~100 rad/s范圍時,受試樣品的η*表現為隨頻率增加呈增加趨勢,類似剪切變稠,內部結構重整。η*沿直線下降可以認為產生了剪切變稀溶液,歸因于無規卷曲和纏結分子被打開,而低頻率振蕩過程因纏結未打開而不會出現剪切變稀現象[22]。
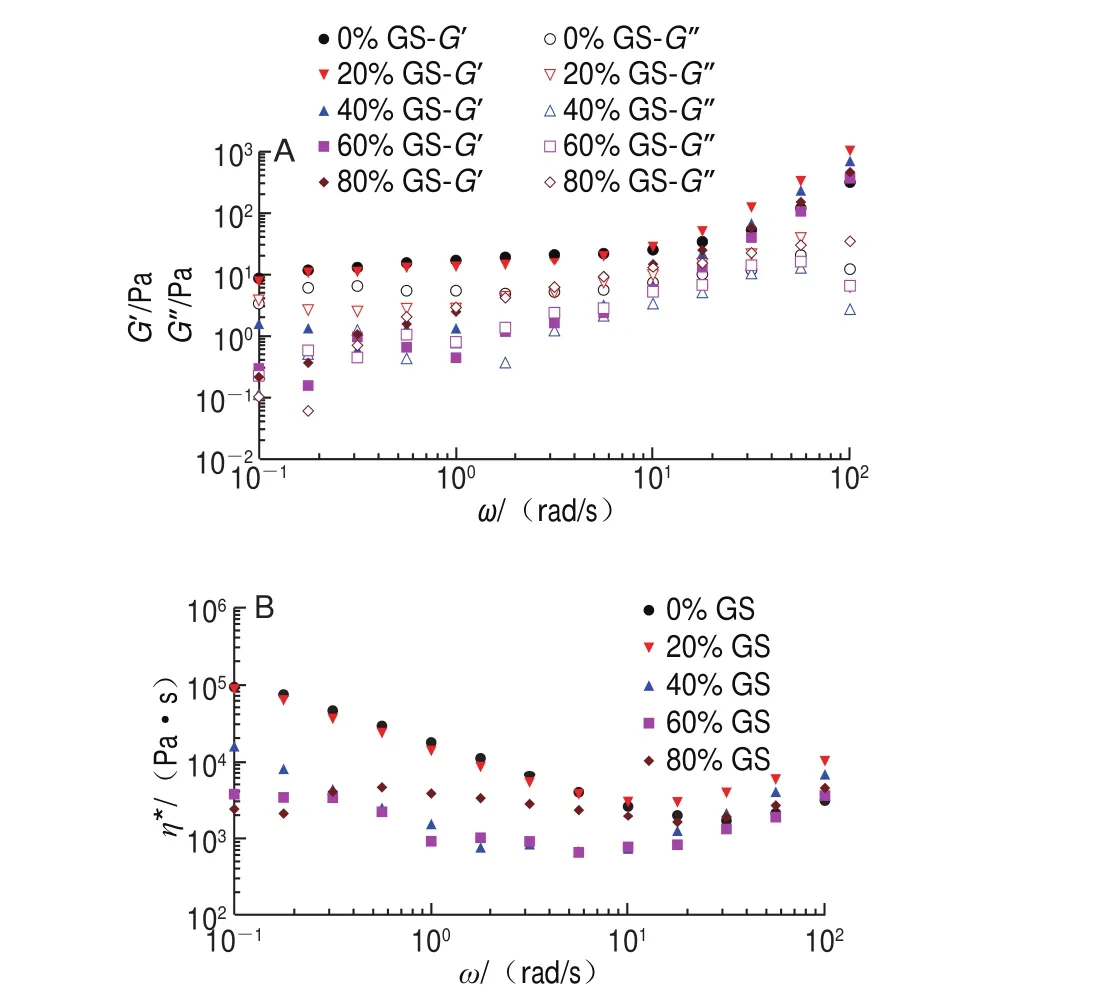
圖 9 明膠/變性淀粉共混體系升溫程序末黏彈模量-ω(A)及η*-ω(B)頻率掃描曲線Fig. 9 G’ and G” versus ω (A) and η* versus ω (B) curves of GL/MS blends at the end of heating stage
2.3 葡萄糖漿共溶質場中共混體系非等溫階段相轉變過程動力學分析結果
本研究采用的明膠/變性淀粉模型體系中,明膠組分直接對凝膠行為起作用,變性淀粉組分為低黏度酸化淀粉,主要利用變性淀粉占位效應使明膠于較低質量分數下形成凝膠。雖然明膠/變性淀粉共混體系流變學性質受到變性淀粉及葡萄糖漿的影響,但主要由明膠組分主導,圖10為變性淀粉及葡萄糖漿分子擁擠環境下明膠溶膠-凝膠相互轉變機理解析模式。
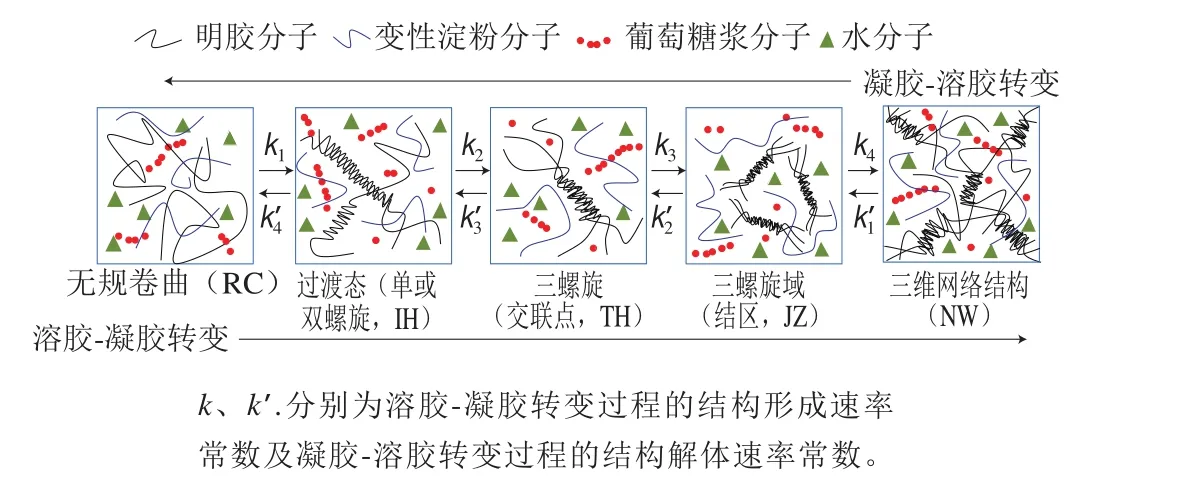
圖 10 明膠/變性淀粉共混體系溶膠-凝膠及凝膠-溶膠轉變機理示意圖Fig. 10 Schematic presentation of the sol-gel and gel-sol transition of GL/MS blends
2.3.1 溶膠-凝膠相轉變動力學解析
根據圖10明膠/變性淀粉共混體系的相轉變特征,提出簡化后的溶膠-凝膠相轉變動力學方程模型(式(8))。
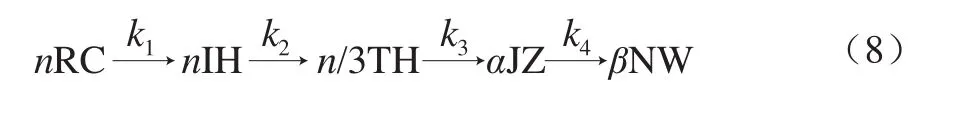
式中:RC為無規卷曲;IH為過渡態螺旋結構;TH為三螺旋結構,即交聯點;JZ為三螺旋交聯區,即結區;NW為三維網絡結構;n、α、β分別為有效結構單元數;k1~k4分別為各轉變階段的結構形成速率常數。
第I階段對應溫度區間為[Ti,Tig],共混體系處于高分子濃溶液狀態,其中水為溶劑,葡萄糖漿為共溶質,明膠分子及變性淀粉分子呈無規卷曲狀態,高分子鏈之間可接觸、交疊,但尚無TH形成,體系可流動。明膠分子主要以RC及IH形式存在,且無規卷曲明膠分子濃度高于單螺旋或雙螺旋過渡態分子濃度,即cRC>cIH;第II階段對應溫度區間為[Tig,Tg],對應高分子體系狀態為交聯態區(或稱為預凝膠區),共混體系明膠高分子狀態包括RC、IH、TH及JZ。明膠各種分子狀態濃度排序為:cRC>cIH>cTH>cJZ>cNW,隨溫度下降,明膠三螺旋聚集體密度的增加,G’呈逐漸增加趨勢[23]。第III階段溫度區間為[Tg,Tcg],對應高分子體系狀態為凝膠前區,4 種結構狀態均存在,隨溫度降低共混體系中cRC不斷降低,單股螺旋增量(ΔcIH)逐漸升高,并在Tcg時達到最大值,cTH及cJZ逐漸升高,始終保持cIH>cTH>cJZ。第IV階段為凝膠后區,溫度區間為[Tcg,Tmg],ΔcIH逐漸降低,ΔcTH及ΔcJZ逐漸升高,但ΔcTH>ΔcJZ,直至ΔcTH達最大值,此刻凝膠化速度達vgmax。第V階段溫度區間為[Tmg,Te],既有明膠三螺旋聚集體不斷轉化為結區結構組分,ΔcTH不斷降低,表現為vg降低,凝膠化網絡不斷強化,G’增加。研究人員提出明膠溶液體系發生溶膠-凝膠轉變的機理存在較大差異,Djabourov等[24]提出明膠的凝膠過程主要涉及兩個過程,首先隨溫度下降起始階段明膠的三螺旋量以指數方式快速增加,第二階段三螺旋量以對數方式增長。Bohidar等[25]提出明膠溶膠-凝膠轉變過程存在3 個明顯特征區域,首先是單體聚集,然后是無規-單股螺旋轉變,緊接著發生單螺旋向三螺旋轉變,并最終形成凝膠。
圖11為未添加葡萄糖漿共混體系Ea求解過程擬合曲線示例,其余濃度葡萄糖漿共混體系Ea曲線趨勢與此相同。由圖11可知,溶膠-凝膠轉變及凝膠-溶膠轉變過程均在兩個不同的溫度范圍(高溫區及低溫區)分為兩個階段反應。溶膠-凝膠轉變及凝膠-溶膠轉變過程高溫區Ea均明顯高于低溫區,文獻[9]發現類似現象。其中溶膠-凝膠轉變過程對應解析圖中的分解點溫度為18.5 ℃(以對照組為例,與Tcg接近),高溫階段對應Ti~Tig~Tcg階段,即對應解析圖(圖2)中I、II及III階段,三螺旋過渡態形成階段需克服更高Ea,此階段k1、k2比較接近。而低溫階段對應IV及V階段,即[Tcg,Te],轉變快慢取決于明膠三螺旋聚集形成的形成速率,此階段k3、k4比較接近。高溫區Ea高于低溫區,表明高溫區明膠三螺旋形成量少的前提下,要形成網絡需克服的能壘更高,低溫區Ea低表明此階段容易形成網絡結構,因為低溫區共混體系中易形成更多的三螺旋聚集物。
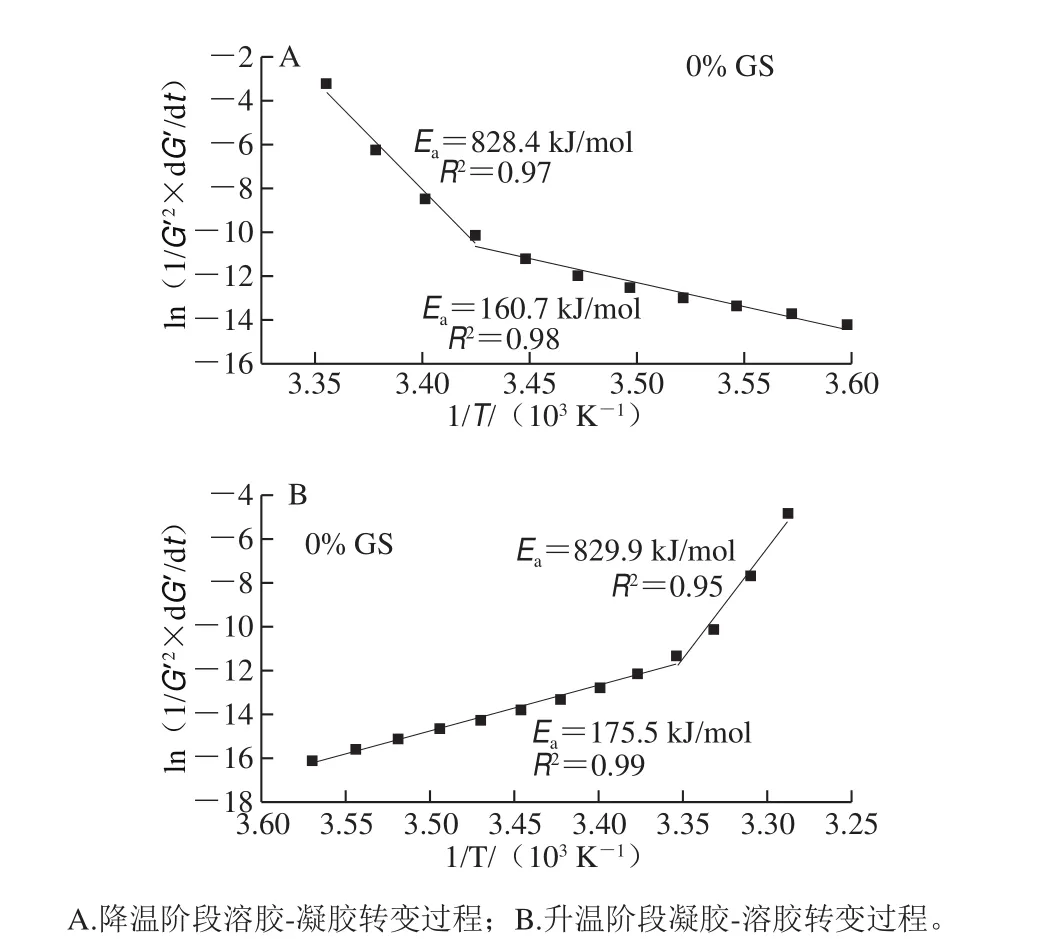
圖 11 明膠/變性淀粉共混體系G’隨溫度變化的Arrhenius擬合曲線Fig. 11 Arrhenius fitting curves for G’ versus T cures of GL/MS blends
2.3.2 凝膠-溶膠相轉變過程動力學解析
根據圖10明膠/變性淀粉共混體系的相轉變特征,凝膠-溶膠相轉變動力學過程可簡化為式(9)。
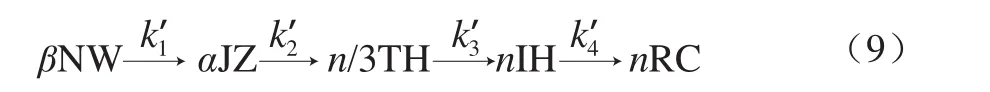
式中:k’1~k’4分別為各轉變階段的結構解體速率常數。
第I階段溫度區間為[Ti,Tis],體系為凝膠態,明膠分子以有序聚集TH形式存在于JZ中,后者構成三維網絡結構,無規卷曲基本不存在。此過程溶膠化速率不斷增加直至最大值(vsmax),伴隨預解離JZ增量降低,但預解離聯結區量增加,部分聯結區解離形成預解離三螺旋,三螺旋含量逐漸相應增加。第II階段溫度區間為[Tis,Tcs],體系處于凝膠結構失穩前區,G’、vs繼續下降,αs正向加速增加至最大值Tcs處,Tcs處三螺旋過渡態開始解螺旋。Nishinari等[26]提出“拉鏈模型”以解釋熱可逆凝膠-溶膠轉變過程,他們認為當溫度升至分子鏈段從拉鏈締結區釋放時,通過拉開分子拉鏈而觸發凝膠-溶膠轉變,與本研究結果類似。第III階段溫度區間為[Tcs,Ts],體系為凝膠態,溫度驅動力減弱,處于凝膠結構失穩區,G’、vs均降低,αs正向加速降低,伴隨對應微區聯結區降低三螺旋量繼續降低,單股螺旋量增加,自由卷曲鏈開始形成,直至微區聯結區解離完成且三維網絡結構骨架(晶種)解螺旋完畢,對應G’=G”,此后三維網狀結構不復存在。拉鏈模型中提出凝膠-溶膠轉變過程部分鏈段構型發生變化后,從局部區域開始逐漸擴散,只要有足夠多的螺旋聚集存在就足以維持三維網絡結構不致于崩塌[27]。第IV階段溫度區間為[Ts,Tms],體系為溶膠態,G’、vs均降低,αs正向加速繼續降低,三螺旋含量繼續降低,單螺旋增量逐漸減少,單股螺旋含量增加,自由卷曲鏈加速形成,自由卷曲鏈增速大于單股螺旋,且越來越大,形成剪刀差效應,直至臨界點Tms三螺旋全部消失,體系中以自由卷曲和單螺旋存在。第V階段溫度區間為[Tms,Te],隨溫度進一步升高,共混體系高分子自由卷曲程度增大,高分子相互作用以纏結、互穿為主,呈濃溶液狀態,流變學上體現為G”>G’,宏觀和微區均不存在TH、IH及三螺旋過渡態。研究人員發現卡拉膠凝膠-溶膠轉變過程存在類似現象[28]。
凝膠-溶膠轉變過程對應解析圖(圖7)中的分解點溫度為25.06 ℃(以對照組為例,與Tcs值接近)。低溫階段對應[Ti~Tcs]溫度段(解析圖中I及II階段),受控于預解離三螺旋形成速率,只需克服更低的Ea,此階段k’1、k’2比較接近。高溫階段對應[Tcs~Te]溫度段(解析圖中III、IV及V階段),受控于三螺旋過渡態形成,需要克服更高能壘,此階段k’3、k’4比較接近。Silva等[29]研究高甲氧基果膠/蔗糖復合體系時也發現高溫區Ea明顯高于低溫區。共混體系凝膠動力學方程受試樣的熱處理歷史影響明顯,溫度較高時Ea較大,主要因為纏結高分子與未纏結高分子之間存在較高的能壘,很難形成連續的網絡結構,而溫度較低區間的Ea較小,分子鏈之間容易發生聚集,并最終形成網絡結構[30]。
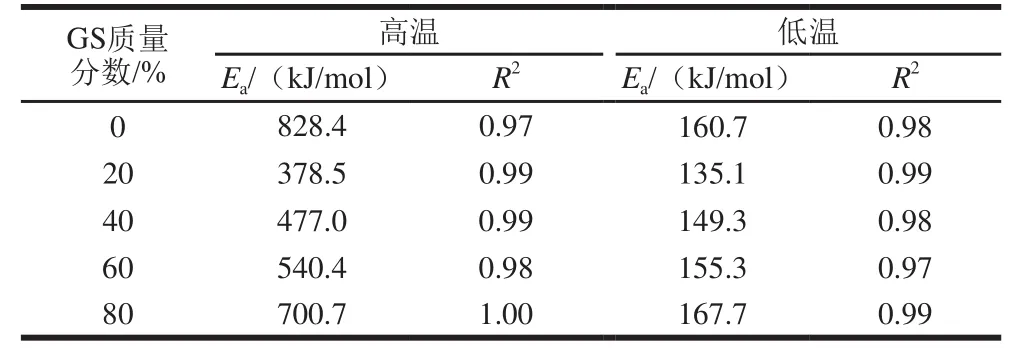
表 6 明膠/變性淀粉共混體系溶膠-凝膠轉變過程凝膠Ea及相關系數特征參數Table 6 Characteristic parameters of Ea and R2during sol-gel transition
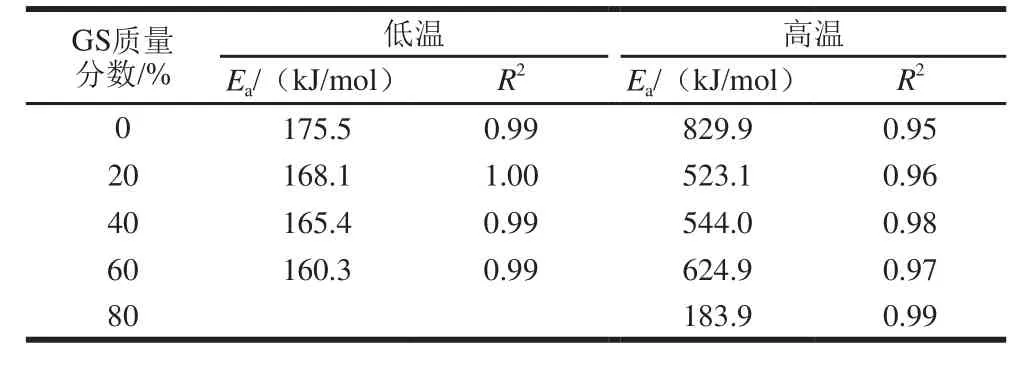
表 7 明膠/變性淀粉共混體系凝膠-溶膠轉變過程溶膠Ea及相關系數特征參數Table 7 Characteristic parameters of Ea and R2during gel-sol transition
表6、7為降溫及升溫過程的Ea擬合結果,反映溶膠-凝膠及凝膠-溶膠轉變過程。與對照組相比,20%葡萄糖漿共溶質存在時,高、低溫區Ea分別由828.4、160.7kJ/mol降低至378.5、135.1kJ/mol,降低幅度分別達54.3%及15.9%,隨葡萄糖漿質量分數增加,均出現先降低后增加趨勢,但總體都低于對照樣,可能因為葡萄糖漿共溶質存在而不利于網絡結構形成且隨共溶質質量分數增加而增加所致。葡萄糖漿質量分數對凝膠-溶膠轉變低溫區Ea影響較小。隨葡萄糖漿質量分數增加,除添加80%葡萄糖漿樣品外,其余樣品高溫區的Ea呈先降低后升高趨勢,三螺旋解聚集阻力隨葡萄糖漿共溶質質量分數增加而上升,但添加80%葡萄糖漿明膠/變性淀粉高溫區Ea發生驟降,可能原因是體系中形成的明膠三螺旋聚集鏈短且數量少。
3 結 論
本研究提出一種依據流變學方法劃分葡萄糖漿共溶質場中明膠/變性淀粉溶膠-凝膠轉變及凝膠-溶膠轉變過程的方法,溶凝轉變及凝溶轉變過程均存在4 個特征溫度值,依據這4 個特征溫度值可將相轉變過程劃分為5 個階段,每個階段具有明顯的物理意義,其高分子存在及運動形式存在明顯差別,流變性質及結構化速率具有明顯差別。明膠/變性淀粉溶膠-凝膠轉變及凝膠-溶膠轉變過程G’、G”隨葡萄糖漿共溶質質量分數增加明顯降低,明顯弱化了凝膠結構。隨葡萄糖漿質量分數(0%~60%)增加,共混體系相轉變過程凝膠點溫度(Tg)呈上升趨勢,而溶膠點溫度(Ts)呈降低趨勢,同時相轉變過程及同溫度點G’、相轉變平均速率、即時速率及加速度呈降低趨勢,表明葡萄糖漿共溶質的存在弱化了共混體系凝膠,且隨葡萄糖漿質量分數增加而加劇。降溫及升溫相轉變過程均存在兩個Ea,高溫階段的Ea均高于低溫階段,隨葡萄糖漿質量分數增加,共混體系相轉變過程高溫區Ea呈先降低后增加趨勢,但低溫區Ea不存在明顯變化。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
