SVIP 通過調節自噬抑制CCl4誘導的肝纖維化的研究
2020-03-14 02:55:26余光銀陶麗麗殷曉敏陳耀麗王柳
中國現代藥物應用 2020年3期
余光銀 陶麗麗 殷曉敏 陳耀麗 王柳
肝纖維化是一種常見的病理狀態,其中肝星狀細胞(HSC)被激活,然后細胞外基質(ECM)蛋白積聚,通常與由感染、藥物、代謝紊亂或自身免疫失衡引起的慢性肝病相關。肝纖維化如果控制不好,將導致不可逆的肝硬化,甚至是肝細胞癌[1,2]。到目前為止,除了去除潛在的病因或肝移植外,還沒有有效的臨床療法來抑制肝纖維化的病理進展。除此之外,研究人員過分關注抑制肝星狀細胞的活化,而不是保護肝臟的功能。畢竟,持續的肝實質細胞死亡對于引發瘢痕形成至關重要[3,4]。因此,需要研究肝纖維化的分子基礎,并開發一種保護實質細胞和逆轉肝纖維化的新治療方法。自噬是一種關鍵的細胞內途徑,破壞的細胞器和受損蛋白質被降解,為真核細胞中的細胞穩態提供能量。自噬途徑通過保守的基因產物的幾個階段進行。在誘導(營養物剝奪或饑餓)后,雷帕霉素靶蛋白復合物1(mTORC1)的抑制激活ULK1/ 2-Atg13-Atg101-FIP200 復合物(Atgs,自噬相關基因),其啟動分離膜或形成噬菌體。自噬的啟動還伴隨著Vps34-Vps15(p150)-Beclin1 復合物的激活。刺激Beclin1 復合物產生磷脂酰肌醇-3-磷酸(PI3P),其促進自噬體膜成核。自噬體延伸需要Atg5-Atg12 和微管相關蛋白1 輕鏈3/酵母自噬相關基因8(LC3/ Atg8)綴合系統[5,6]。此外,LC3 參與選擇性轉運自噬底物蛋白62(p62)/ SQSTM1 和NBR1 等蛋白質,這些蛋白質含有特殊的LC3 相互作用區域基序,用作細胞結構隔離的適配器,如線粒體,蛋白質聚集體和其他細胞結構。GTP 酶Ras 相關蛋白7(Rab7)需要完成與溶酶體融合的自噬體階段。在最后階段,溶酶體酸水解酶降解自噬體內容物,并釋放自溶酶體的內容物用于代謝回收。自噬在調節脂肪形成中起關鍵作用,與脂肪變性和肝纖維化有關。它可以通過脂質減少脂滴。否則,長期脂質負荷可能在體外和體內改變膜脂組成并減少自噬體和溶酶體的融合[7]。因此,在肝臟中過量脂質抑制自噬可能導致肝細胞中脂滴積聚。然而,據報道,活化的自噬通過降解脂滴為肝星狀細胞的活化和增殖提供能量。自噬通過降解膠原蛋白來抑制纖維化。自噬的激活降解小鼠肝臟中的Ⅰ型膠原蛋白,減少氧化應激,并抑制炎癥以抑制纖維化。它還可以保護肝細胞免受凋亡。因此,自噬在肝星狀細胞活化和從脂肪變性到纖維化的過程中的作用是有爭議的。
1 資料與方法
1.1 方法 在饑餓模型中,在對照組(禁食)中,對15 只7 周齡小鼠進行4 h 禁食,1 h 再喂養的模式,之后立即處死。在實驗組(肝纖維化)中,對15 只小鼠注射1 ml/kg 的50%CCl4橄欖油2 次/周,持續8 周,對照組注射相同劑量的橄欖油8 周。對于肝纖維化和禁食模型中,注射CCl4或橄欖油,將處理過的大鼠禁食1 h。之后收集肝組織和血清進行分析。在饑餓期間,這些動物可以獲得飲用水。使用三唑試劑從細胞顆粒或新鮮組織中提取總核糖核酸。如前所述,通過MTT 分析評估細胞活力。經過不同的處理(DMSO 或0.05%CCl4)和不同的培養時間(0、24、48、72 h),在測試波長和參考波長為570、630 nm 的掃描孔微培養板讀取器中讀取孔的吸光度。HepG2細胞在4℃下用10%甲醛固定30 min,用BODIPY493/503 染色15 min。用磷酸緩沖液(PBS)洗滌后,細胞核用DAPI 染色20 min。熒光顯微鏡檢測脂滴和自噬體,進行圖像采集和圖像分析。
1.2 統計學方法 采用GraphPad Prism 第6 版統計學軟件處理數據。數據表示為至少三個獨立實驗的平均值標準劃分,兩組以上數據比較進行單向方差和事后多重分析。P<0.05 表示差異有統計學意義。
2 結果
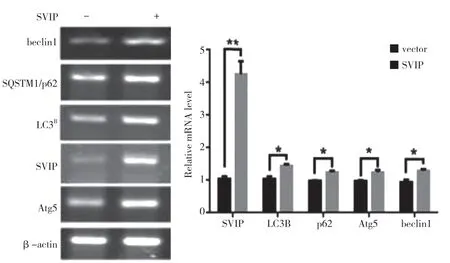
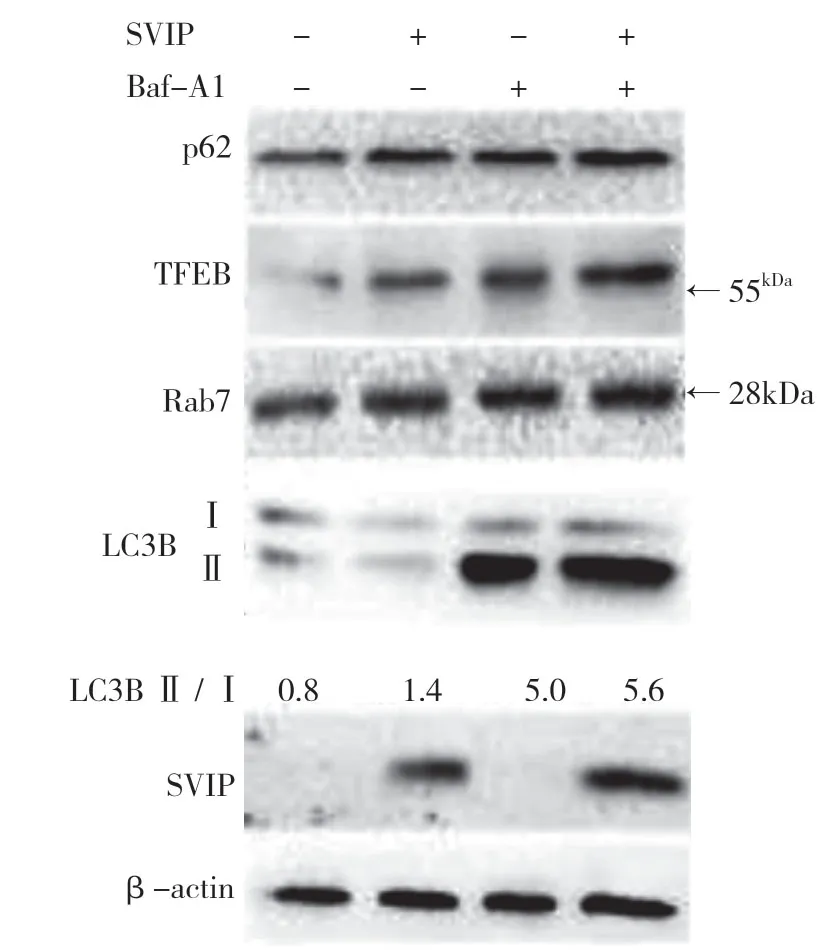
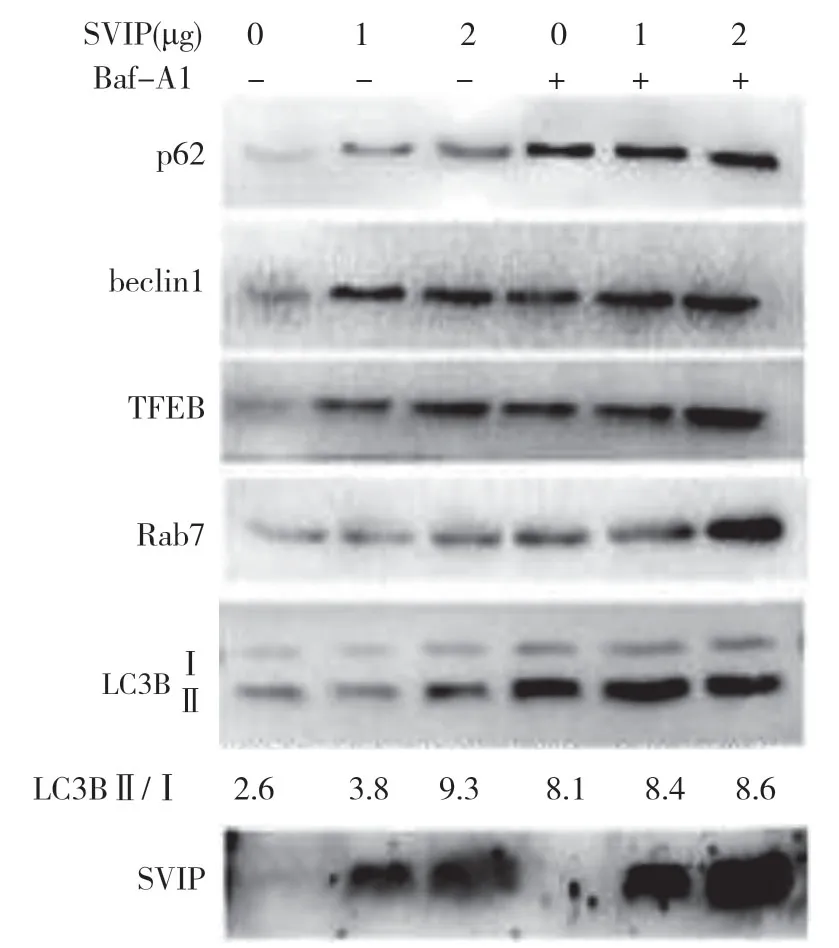
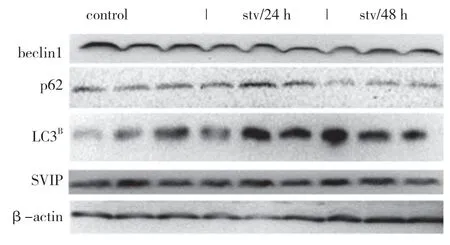
自噬相關基因的表達,包括LC3(Atg8 的哺乳動物同源物)、酵母自噬相關基因6(Atg6)、酵母自噬相關基因5(Atg5)和p62/SQSTM1,在蛋白質和基因水平上均與SVIP 過度表達呈正相關(見圖a,b)。與對照組相比,它們在SVIP 細胞中的表達減少(見圖e,f)。為了評估自噬通量,LC3-Ⅱ/LC3-Ⅰ通過巴非霉素A1 計算,巴非霉素A1 中和溶酶體的酸堿度并阻斷自噬體-溶酶體融合(見圖c,d)。轉錄因子EB 的表達由于SVIP 過度表達而以劑量依賴的方式增加(見圖c,d),這解釋了SVIP 在轉錄水平上增加自噬相關基因(Atgs)和p62 的表達(見圖b,f)。以上所有證據表明SVIP 促進了肝癌細胞的自噬。
饑餓可以在體外和體內激活自噬。此外,SVIP 增強了饑餓誘導的自噬。但是,饑餓是否會增加肝臟或肝癌細胞中的SVIP 表達仍然未知。盡管LC3、p62、Beclin1 和Atg5 的表達因營養缺乏而顯著改變,但SVIP 表達對HepG2 細胞的饑餓不敏感,也不在肝臟中,在肝臟組織中禁食24、48 h 后,SVIP 仍然是不可檢測的(見圖g,h,i)。同樣,SVIP 表達不受小鼠大腦饑餓的影響(見圖j)。
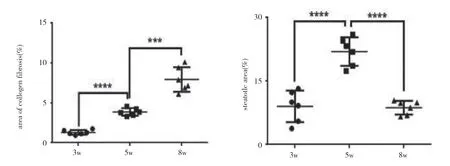
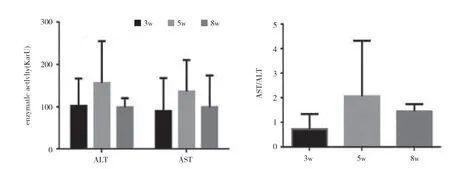
大鼠接受CCl4處理分別注射3、5、8 周。在肝纖維化的發展過程中,大鼠肝臟的病理特征發生了顯著變化。最初,膠原纖維的區域和脂肪變性區域被觀察到。纖維化大鼠肝臟顯示不同程度的損傷:輕度,中等,嚴重,對應于3、5、8 周CCl4分別治療。圖表顯示,倒V 形曲線以及脂滴的大小。在中度纖維化大鼠肝臟中達到頂點(見圖k,l,m,n)。此外,采用抗形狀記憶合金α 的天門冬氨酸氨基轉移酶(AST)/丙氨酸轉氨酶(ALT)生化分析測定肝功能損傷和肝纖維化的嚴重程度分別證實了進展中的肝纖維化,此外,分析相關基因產物的表達,蛋白質印跡試驗和逆轉錄酶聚合酶鏈反應來評估自噬。SVIP 和LC3 的表達在第5 周比第3 周增加,然后在第8 周下降(見圖o)。最后,SVIP 和基因水平在第3~8 周也呈倒V 型曲線。在CCl4期間治療自噬活性在5 周達到頂峰,然后在8 周下降。這些數據表明,SVIP 的表達與自噬活性以及纖維化大鼠肝臟中脂滴的大小密切相關。
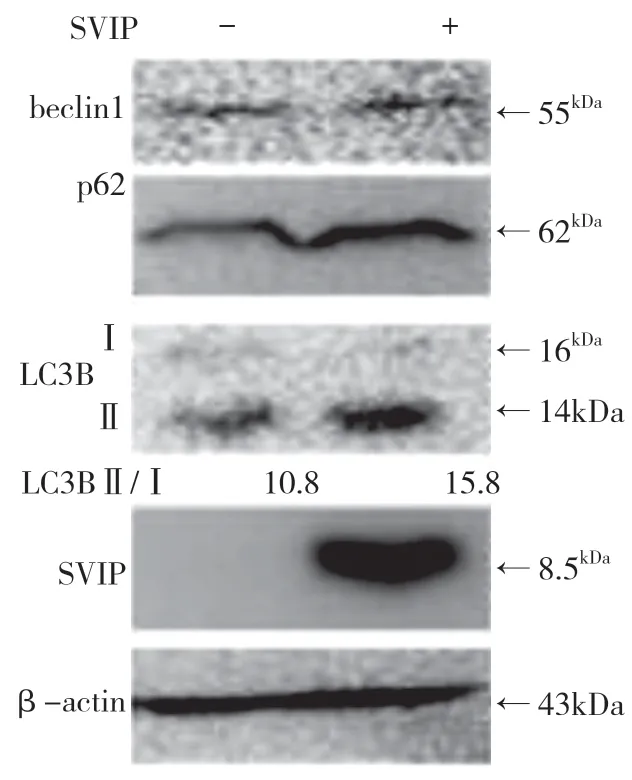
圖a
圖b
圖c
圖d
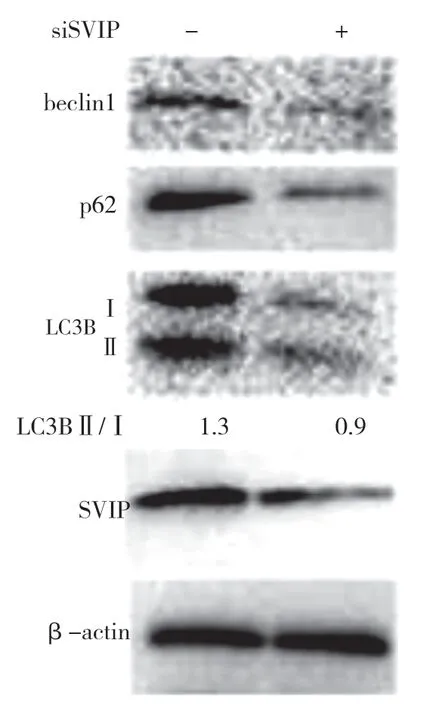
圖e
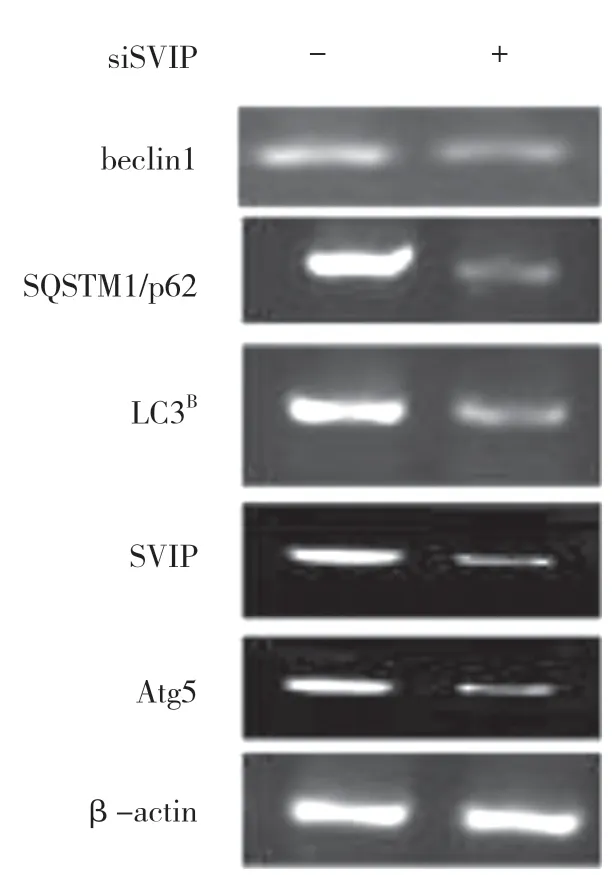
圖f
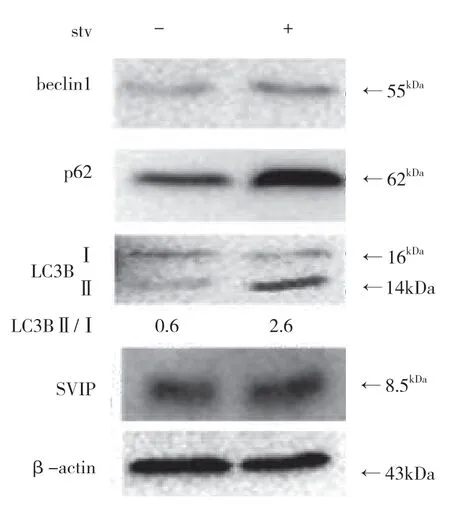
圖g
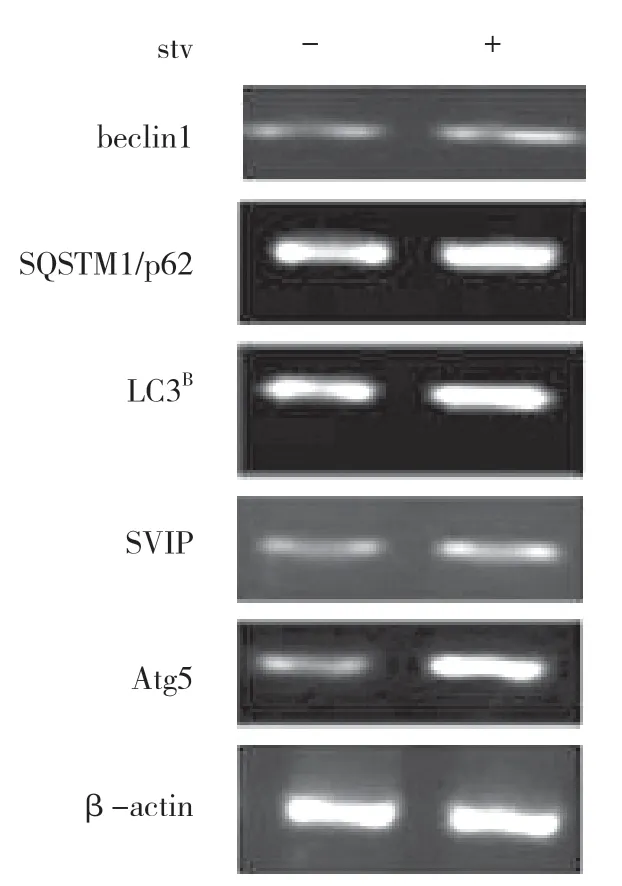
圖h
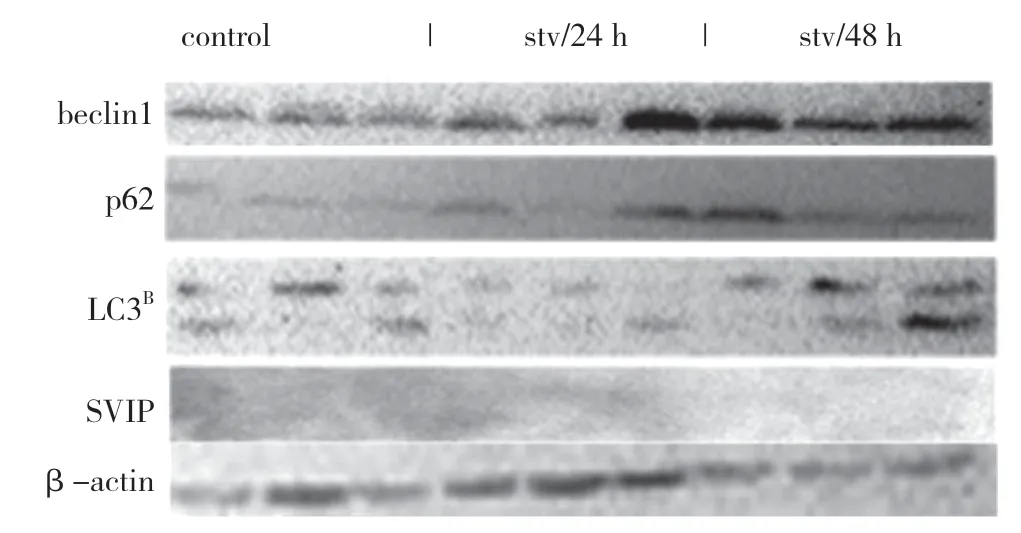
圖i
圖j
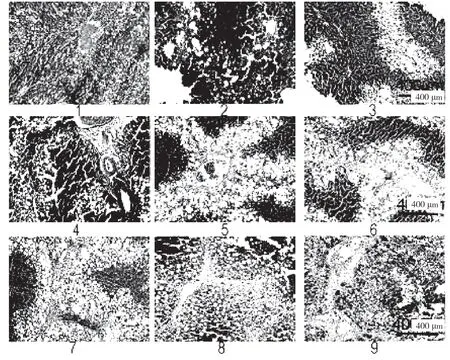
圖k
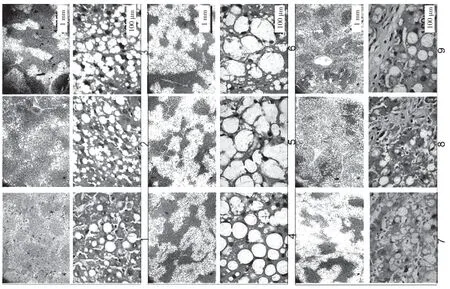
圖l
圖m
圖n
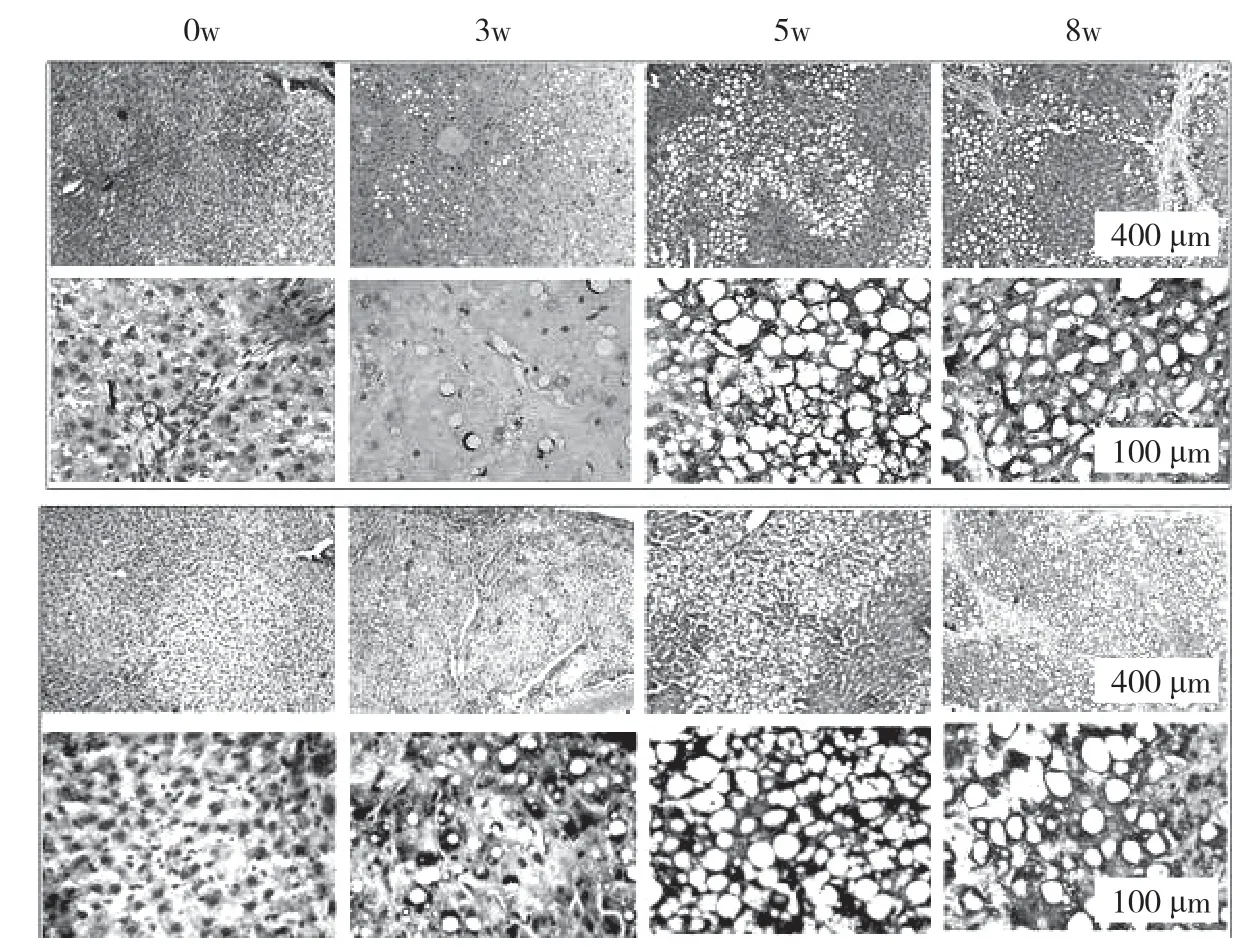
圖o
3 討論
SVIP 保護肝細胞的機制可能包括減少脂肪變性肝臟中脂肪酸的積累和增強抗氧化作用。最初,研究表明肝細胞由于CCl4刺激導致脂肪酸的積累和脂質過氧化引起了細胞毒性。因此,SVIP 可以調節自噬,減少脂質的積累[8,9]。此外,SVIP 有一個含纈酪肽蛋白(VCP)相互作用基序,而泛素連接酶羥甲基戊二酰輔酶A 還原酶降解蛋白1(Hrd1)和STUB1/CHIP 依賴它們的VCP 結合基序降解底物。相互作用基序和結合基序都能與VCP 脫氫酶第一亞單位(ND1)結構域結合,ND1 與相互作用基序的親和力高于VBM25。因此,SVIP 的增加競爭性地抑制了Hrd1 和STUB1/CHIP 與VCP 的結合,并減少了它們的底物降解。Nrf2 和TFEB,Hrd1 和STUB1/CHIP 的底物,是調節與抗氧化應激和溶酶體生物發生相關的基因的轉錄因子[10-12]。SVIP 的表達在CCl4大鼠肝臟和肝癌細胞中增多。該機制可能與泛素-蛋白酶體途徑的激活有關。一些研究表明,內質網應激增加了SVIP,激活了泛素蛋白酶體途徑。當CCl4導致肝細胞內質網應激,內質網相關降解和SVIP 表達增加。自噬被SVIP 激活以保護肝細胞。如果SVIP被耗盡,自噬被抑制。同時,HepG2細胞對CCl4毒性更敏感。自噬與肝脂肪變性和纖維化密切相關[13-15]。然而,從脂肪變性到纖維化過程中的自噬動力學鮮有報道。本研究結果表明自噬隨著肝脂肪變性的發展而激活,然后一旦發展成纖維化就失活。這里,在CCl4中的治療模型,激活自噬或SVIP 表達可減輕肝纖維化。隨著激活的自噬,脂滴的體積不斷增加,而在肝癌細胞中SVIP 過度表達或饑餓也顯示出增大的脂滴。
綜上所述,SVIP 的表達與CCl4形成過程中的自噬水平密切相關,且誘導肝纖維化。SVIP 能誘導自噬和延緩肝纖維化。研究SVIP 激活自噬的分子機制具有十分重要的意義,為今后抗纖維化提供了新的理論依據。
