金黃色葡萄球菌SaeP蛋白表達條件的優化
2020-03-23 11:42:48劉佳璐鄭維范若辰權春善
中國乳品工業 2020年1期
關鍵詞:優化
劉佳璐,鄭維,范若辰,權春善
(1.大連民族大學a.生命科學學院;b.生物技術與資源利用教育部重點實驗室,遼寧大連116600;2.大連理工大學生命科學與技術學院,遼寧 大連116024)
0引言
金黃色葡萄球菌(Staphylococcusaureus),是導致奶牛乳房炎的主要病原菌之一[1-4],其中的SaeRS雙組分信號轉導系統調控多種毒力因子的表達,由傳感器組氨酸激酶SaeS和響應調節劑SaeR以及脂蛋白SaeP、膜蛋白SaeQ等兩種輔助蛋白組成。SaeP與SaeQ以及SaeS共同形成三元復合物[5-6],該三元復合物可以激發SaeS磷酸酶活性,對SaeR蛋白進行磷酸化,使下游的Sae操縱子表達,最終激發多種毒力因子生成。SaeP蛋白表達量較低,限制了Sae系統的深入研究。本課題以優化SaeP蛋白表達條件為目的,通過改變重組載體構建方式和誘導溫度等方式,確定最優SaeP表達條件,提高SaeP蛋白的表達量,推動SaeRS系統的整體研究,為研究金黃色葡萄球菌致病機理提供理論依據。
1實驗
1.1材料與試劑
1.1.1菌種和質粒
金黃色葡萄球菌、p ET-28a、Tuner(DE3)、C43(DE3)、BL21(DE3)-pLysS以及BL21(DE3)-RIL均由本實驗室保存。含有saep基因的質粒pET-28a-saep由本實驗室構建。Trans5αChemically Competent Cell(目錄號:CD201)和BL21(DE3)Chemically Competent Cell(BL21(DE3)目錄號:CD601)。
1.1.2主要試劑
Nco I,Xho I,T4 DNA Ligase,Dpn I,DL2,000 DNA Marker,Blue Plus II Protein Marker(14-120 ku),WB封閉液(in PBS)、SanPrep柱式DNA膠回收試劑盒,基因組提取試劑盒,質粒提取試劑盒,Axyprep PCR清潔試劑盒,配制培養基用酵母提取物、胰蛋白胨,其余試劑均為國產分析純。
1.1.3溶液與培養基的配制
1 mmol/L IPTG的配制:將2.38 g IPTG溶解于水中,定容至100 mL。
LB培養基配方(1 L):胰蛋白胨10 g、酵母提取物5 g、氯化鈉10 g,定容至1 L。配制固體培養基則再加入20 g的瓊脂粉。配制完成后將p H值調節至7.0,使用時根據需要添加抗生素。
SOB培養基配方(1 L):蛋白胨20 g、酵母提取物5 g、氯化鈉0.5 g加入950 mL水溶解后,加入濃度為0.25 mol/L的KCl溶液10 mL,用NaOH調節p H值到7.0,定容至1 L,高壓滅菌20 min。(該溶液使用前加入5 mL滅菌的濃度為0.002 mol/L的MgCl2溶液)。
SOC培養基:將SOB培養基高壓滅菌后,降低溫度到60℃以下時,加入20 mL用0.22μm濾膜過濾除菌的濃度為1 mol/L葡萄糖溶液,即為SOC培養基。
1.1.4 4種感受態細胞的制備
采用氯化鈣法[7]將Tuner(DE3)、C43(DE3)、BL21(DE3)-pLysS以及BL21(DE3)-RIL制作成相應的感受態細胞。
In the first experiment,we demonstrate the performance of the proposed algorithm under different angular distributed functions.The parameters of two ID sources areandThe numberof snapshots is set as T=500 and the Signal-to-Noise Ratio(SNR)is equal to 5 dB.We perform the algorithm in three cases:
1.2方法
1.2.1重組質粒pET-28a-saep的構建
通過酶切酶連的方式重新構建重組質粒p ET-28a-sae p。選取酶切位點為Nco I和Xho I,引物設計序列見表1。重組質粒構建完成后,通過雙酶切的方法對其進行驗證。
通過無限克隆法(Restriction Free Cloning,RF法)構建重組質粒p ET-28a-saep。用RF-Cloning設計引物。引物設計序列見表1。通過PCR儀對目的基因進行擴增,并且將目的基因與質粒pET-28a相連,完成重組質粒pET-28a-saep的構建。通過對重組質粒pET-28a-saep的PCR產物進行電泳驗證。
引物的合成以及檢測結果正確的質粒的測序工作均由上海生工生物工程技術服務有限公司完成。SDS-PAGE分析以及Western blot檢測。通過Western Blot實驗對蛋白進行驗證,再通過SDS-PAGE蛋白電泳將兩種方法構建的重組蛋白進行對比分析。
1.2.3宿主菌的優化
將重組質粒p ET-28a-saep分別轉入大腸桿菌BL21(DE3)、Tuner(DE3)、C43(DE3)、BL21(DE3)-p LysS以及BL21(DE3)-RIL中,均勻涂布于帶有卡那霉素抗性的LB固體培養基上,37℃過夜培養,挑取單菌落,培養10 h,再進行擴大培養,OD 600達到0.6時加入終濃度為0.5 mmol/L的IPTG。30℃誘導8 h后,分別取5種不同宿主的菌液進行超聲破碎后,離心30 min,取上清作為樣品加入進行SDS-PAGE分析。
1.2.4誘導劑濃度的優化
根據本文2.2.2的實驗結果,選擇最適宜的宿主菌BL21(DE3)進行誘導劑濃度的優化。將已經轉入重組質粒pET-28a-saep的BL(DE3)接種LB液體培養基中,培養10 h,再進行擴大培養,至OD 600達到0.6時加入IPTG,至終濃度分別為0,0.5,0.9,1.3和1.5 mmol/L,30℃誘導8 h,分別取5種不同誘導劑濃度的菌液進行超聲破碎后,離心30 min取上清作為樣品進行SDS-PAGE分析。
1.2.5誘導溫度及誘導時間的優化
根據上述2.2.3中的實驗結果,將誘導劑終濃度定為0.5 mmol/L,進行下一步誘導溫度及誘導時間的優化。將溫度按梯度分為16,20,24,28以及30℃(原誘導溫度),誘導時間分為8,12,24 h,分別進行平行實驗,具體誘導溫度與誘導時間的設置如表2所示。
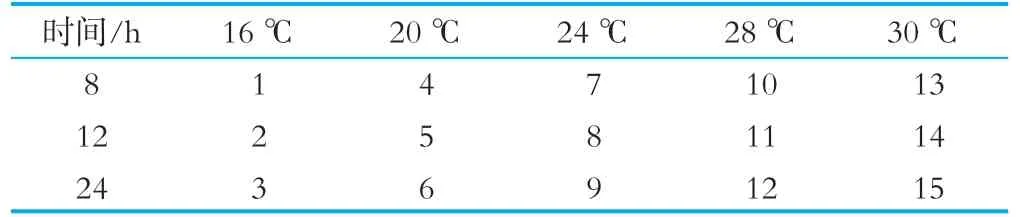
表2誘導時間及溫度設置
按實驗計劃進行活化、轉接、誘導、收菌和破碎,離心后取上清作為樣品進行SDS-PAGE分析。
1.3數據統計
所有實驗重復3次,實驗結果平均值±標準偏差表示。采用Quantity One軟件進行蛋白定量分析,采用2010軟件進行數據分析,采用SPSS 17.0軟件進行差異顯著性分析(P≤0.05)。

表1引物序列
1.2.2構建方式的優化
將兩種方式構建的重組質粒分別轉入BL21(DE3)宿主中,涂布于含卡那霉素抗性的LB固體培養基上,37℃過夜培養,挑取單菌落,培養10 h,將菌液以1%的接種量接種于50 mL的LB液體培養基中,37℃,180 r/min進行培養,OD 600達到0.6時加入終濃度為0.5 mmol/L的IPTG。30℃誘導8 h后,將菌液進行超聲破碎后離心30 min,取上清作為樣品,進行
2結果與分析
2.1重組質粒的構建和驗證
2.1.1酶切酶連法構建重組質粒pET-28a-saep驗證結果
圖1為酶切酶連法構建重組質粒p ET-28a-saep雙酶切鑒定電泳結果圖。由圖1可以看出,重組質粒條帶大小與預期相符,且雙酶切后出現與目的基因條帶大小一致的電泳條帶,質粒測序結果正確,認定酶切酶連法成功構建重組質粒。

圖1酶切酶連法構建重組質粒pET-28a-saep雙酶切鑒定電泳結果
2.1.2 RF法構建重組質粒p ET-28a-saep驗證結果
由圖2可以看出,以重組質粒pET-28a-saep為模板,進行PCR擴增,能夠擴增出單一且分子量大小符合預期的目的條帶。質粒測序結果正確,認定通過RF法成功構建重組質粒。圖2中,1~3為saep基因的PCR擴增產物。
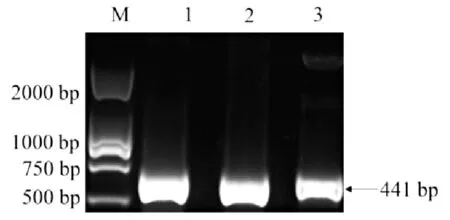
圖2 PCR產物的驗證結果
2.2重組蛋白表達驗證結果
重組質粒pET-28a-saep轉入大腸桿菌BL21(DE3)的誘導產物經SDS-PAGE分析,未添加IPTG誘導的產物與添加IPTG誘導后的產物在大約16 ku位置存在顯著差異。Western blot結果如圖3所示,表達產物能夠與帶有組氨酸標簽的抗體結合,從而顯現出一條清晰的條帶,大小符合預期。
2.3構建方式的優化結果
由圖4(a)可知,RF法與酶切酶連法構建的重組蛋白相對表達量有明顯差異。通過Quantity One和SPSS軟件對其進行分析可知,全菌體蛋白狀態對比下,統計學分析顯示RF法構建全菌體蛋白的相對表達量明顯高于酶切酶連法構建全菌體蛋白的相對表達量,更利于后續純化工作的進行。鎳離子親和層析純化后的酶切酶連法構建重組表達蛋白進一步損失,而RF法構建重組蛋白SaeP的相對表達量大幅提高,所以選擇RF法構建的重組質粒p ET-28a-saep繼續后續實驗的研究。
2.4宿主菌的優化結果
通過Quantity One和SPSS軟件對重組蛋白SaeP的相對表達量進行分析。由圖4(b)可知,通過RF法構建重組質粒p ET-28a-saep以大腸桿菌BL21(DE3)為宿主菌時相對表達量高于其他宿主菌,占菌體蛋白的19.9%,統計學分析顯示以大腸桿菌BL21(DE3)為宿主菌與其他宿主菌相比,具有顯著差異,所以選擇以大腸桿菌BL21(DE3)為宿主菌進行后續實驗。重組蛋白的表達量受基因自身性質、表達載體宿、主菌以及培養條件等多種因素的影響,通過對比不同宿主菌對于重組SaeP蛋白的表達量,以確定較為適合的宿主菌,提高了表達效率[8-11]。
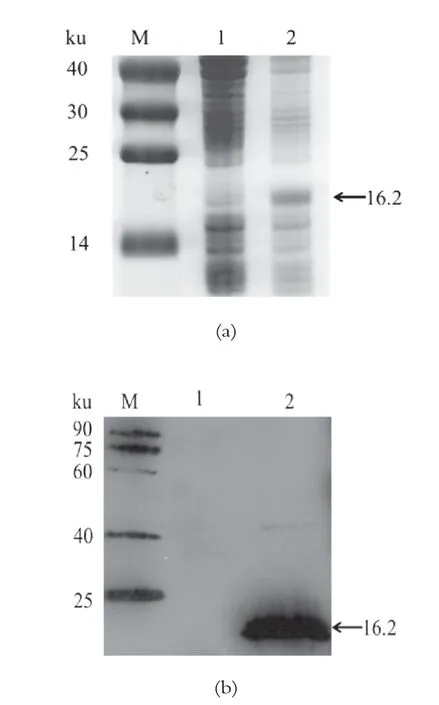
圖3重組表達產物SDS-PAGE蛋白電泳圖及WesternBlot結果
2.5誘導劑濃度的優化結果
通過Quantity One和SPSS軟件對重組蛋白SaeP的相對表達量進行分析,如圖4(c)所示,重組蛋白的相對表達量在IPTG終濃度為0~0.5 mmol/L范圍內,成上升趨勢,當IPTG終濃度達到0.5 mmol/L時重組SaeP蛋白的相對表達量達到峰值,占菌體蛋白的20.16%。誘導劑IPTG的濃度與蛋白表達速率相關,在一定范圍內提高IPTG的濃度會提高蛋白的表達量,但濃度進一步提高后,相對表達量反而有所減少,說明IPTG具有一定的細胞毒性,濃度過高不利于菌株的生長并且抑制細胞活性,所以適當降低IPTG的濃度可能更利于重組蛋白的表達[11-13]。統計學分析顯示濃度為0.5 mmol/L的IPTG誘導重組蛋白相對表達量與其他濃度相比,具有顯著差異,所以在本實驗中,選擇濃度為0.5 mmol/L作為誘導重組蛋白SaeP的誘導劑濃度。
2.6誘導溫度及誘導時間的優化結果
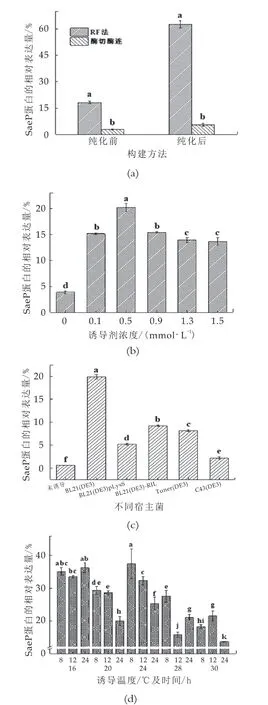
圖4誘導條件優化結果
通過Quantity One和SPSS軟件對重組蛋白SaeP的相對表達量進行分析。由圖4(d)可知,在誘導條件為16℃誘導8 h,16℃誘導12 h,16℃誘導24 h及24℃誘導8 h相對表達量較高,通過對比實驗結果發現低溫誘導條件下重組蛋白表達效果更好。低溫條件下菌體生長速率下降,很大程度上避免了菌體失活與質粒穩定性下降等問題,低溫誘導能夠提高外源蛋白穩定性和可溶性,從而增加了重組蛋白的表達量[14]。重組SaeP蛋白在16℃條件下誘導8 h后,并沒有隨著誘導時間的延長而明顯增加。推測原因是隨著誘導時間的增加,菌體內蛋白酶對重組蛋白的降解量增加,并且誘導時間過長產生的氨基酸會抑制目的蛋白的合成,所以長時間的誘導不利于重組蛋白的表達[15-16]。所以選擇16℃誘導8 h為重組SaeP蛋白的誘導溫度及誘導時間。
3結論
本研究通過改變重組載體構建方式、宿主菌、誘導劑終濃度、誘導時間誘導溫度等,對金黃色葡萄球菌SaeP蛋白的表達條件進行優化,以提高SaeP蛋白的表達量。前期實驗結果表明,無限克隆方法獲得的表達載體在各條件的試驗中,表達量均高于酶切酶連方法所得到的表達載體,因此條件優化部分全部以無限克隆方法得到的表達載體為主要研究對象。成功優化金黃色葡萄球菌SaeP蛋白的誘導表達條件,最佳表達條件為:通過無限克隆法建重組質粒,以BL21(DE3)為轉化宿主,IPTG濃度為0.5 mmol/L,誘導溫度為16℃,誘導時間為8 h,金黃色葡萄球菌SaeP蛋白的最高表達量占總蛋白的35.14%,較原誘導條件提高25.49%,為促進金黃色葡萄球菌的Sae系統對于毒力因子生成研究的提供支持。
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
能源工程(2022年1期)2022-03-29 01:06:28
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
今日農業(2020年16期)2020-12-14 15:04:59
消費導刊(2018年8期)2018-05-25 13:20:08
家庭影院技術(2018年4期)2018-05-09 07:07:41
電子制作(2017年20期)2017-04-26 06:57:45
