二氫楊梅素固體分散體的制備及其體內藥動學研究
2020-03-27 13:43:40范炎峰荊玲范金點鄒夢夢劉寬浩
中成藥 2020年3期
關鍵詞:血漿
范炎峰荊 玲范金點鄒夢夢劉寬浩
(1.黃河科技學院,河南 鄭州450063; 2.廣東藥科大學,廣東 廣州510006; 3.鄭州市第六人民醫院,河南 鄭州450015)
二氫楊梅素主要存在于蛇葡萄科葡萄屬植物中,是一種二氫黃酮醇類化合物,具有明顯的抗炎、抗過敏作用,其機制可能與抑制組胺和白三烯釋放有關[1],并且清除氧自由基也是其發揮抗炎活性的機制之一[2],除此之外,它還具有抗腫瘤、降血脂、降血壓、抗糖尿病、預防心腦血管疾病等多種藥理作用[3-6]。但二氫楊梅素溶解度很差,導致其體內吸收受到極大限制[7],嚴重影響藥效發揮,目前相關制劑學研究有脂質體、自微乳、微囊等[8-10]。
固體分散體是將難溶性藥物在一定條件下高度分散到水溶性高分子材料(如PVP、PEG 等)中形成的一種給藥制劑,可顯著增加溶解度、溶出速率、溶出度[11-12],其制備工藝簡單,輔料安全性高,應用非常廣泛。因此,本實驗將二氫楊梅素制成固體分散體,對親水性高分子材料及其用量進行篩選,采用XRD 技術研究晶型變化,測定溶解度、溶出度,考察體內藥動學,以期為相關口服制劑研究提供參考。
1 材料
1.1 儀器 Agilent 1260 型高效液相色譜儀(配置DAD 檢測器、自動進樣器等,美國Agilent 公司);ME155DU 型電子分析天平[梅特勒-托利多儀器(上海)有限公司];ZNCLBS 型智能數顯加熱磁力攪拌器(上海越眾儀器設備有限公司);D8 ADVANCE 型X 射線粉末衍射儀(瑞士Bruker 公司);MD400-2 型氮氣吹掃儀(浙江杭州奧威儀器有限責任公司);THZ-82 型往返恒溫振蕩器(江蘇金怡儀器科技有限公司);MixPlus 型漩渦混合儀(合肥艾本森科學儀器有限公司)。
1.2 試劑與藥物 二氫楊梅素對照品(批號Y25A6K1,純度98.6%,上海源葉生物科技有限公司);二氫楊梅素原料藥(批號20171125P,純度>98%,湖北遠成賽創科技有限公司孝南分公司);芹菜素對照品(批號P0587,純度99.4%,成都瑞芬思生物科技有限公司)。PEG 4000(批號P161010,海安國力化工有限公司);聚乙烯吡咯烷酮 K30(PVP K30,批號25000240379,亞什蘭集團公司);泊洛沙姆188(F68,批號20160512,湖北鴻運隆生物科技有限公司)。SD 大鼠,雌雄兼具,體質量約300 g,購自河南省實驗動物中心,動物生產許可證號SCXK(豫)2016-0002。
2 方法與結果
2.1 二氫楊梅素含有量測定
2.1.1 色譜條件 Waters C18色譜柱(250 mm×4.6 mm,5 μm);流動相乙腈-0.1% 磷酸(45∶55);體積流量1.0 mL/min;柱溫30 ℃;檢測波長290 nm;進樣量20 μL。
2.1.2 線性關系考察 精密稱取干燥的二氫楊梅素對照品20 mg,轉移至50 mL 量瓶中,乙腈超聲溶解,放置0.5 h 后流動相定容,得0.4 mg/mL 貯備液,流動相逐步稀釋,得到40、20、10、1.0、0.05 μg/mL 對照品溶液,在“2.1.1”項色譜條件下進樣測定。以溶液質量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸,得方程為Y=31.339X+2.954 6(r=0.999 9),在0.05~40 μg/mL范圍內線性關系良好。
2.1.3 方法學考察 取高、中、低質量濃度(40、20、0.05 μg/mL)對照品溶液,在“2.1.1”項色譜條件下進樣測定6 次,測得峰面積RSD 均小于0.41%,表明儀器精密度良好。取10 mg 固體分散體于50 mL 量瓶中,20 mL 乙腈超聲處理后流動相定容至50 mL,于0、1、2、3、4、5 d 在“2.1.1”項色譜條件下進樣測定,測得峰面積RSD 為1.05%,表明溶液在5 d 內穩定性良好。取固體分散體適量,平行制備6 份溶液,在“2.1.1”項色譜條件下進樣測定,測得峰面積RSD 為1.33%,表明該方法重復性良好。精密稱取含有量已知的固體分散體適量,加入不同質量濃度對照品溶液,置于50 mL 量瓶中,乙腈超聲處理后流動相定容,在“2.1.1”項色譜條件下進樣測定,測得平均加樣回收率為99.11%,RSD 小于1.48%。
2.2 體外溶出度試驗 取待測樣品適量(以二氫楊梅素計30 mg),加入3 mL 蒸餾水,置于活化后的透析袋中[13](截留分子量8 000~10 000 Da),溶出儀轉速100 r/min,溫度37 ℃,釋放介質為900 mL經脫氣處理的蒸餾水。于預定時間取樣3 mL,同時加入3 mL 空白溶出介質,溶液經0.45 μm微孔濾膜過濾,在“2.1.1”項色譜條件下進樣測定,計算含有量。
2.3 固體分散體制備 稱取二氫楊梅素20 mg、載體材料(PVP K30、PEG 6000、F68)適量于圓底燒瓶中,加入50 mL 無水乙醇,45 ℃水浴下磁力攪拌均勻,放置4 h 至澄清,減壓旋蒸除去有機溶劑,殘留物置于45 ℃真空干燥箱中干燥24 h,即得,粉碎后過24 目篩備用。
2.4 處方篩選
2.4.1 載體材料 固定二氫楊梅素與載體質量比1∶8,考察PVP K30、PEG 6000、F68 對二氫楊梅素溶出速率、溶出度的影響,結果見圖1。由此可知,PVP K30 作為載體材料時溶出速率及240 min內累積溶出度均高于F68、PEG 6000,故選擇其作為載體材料。
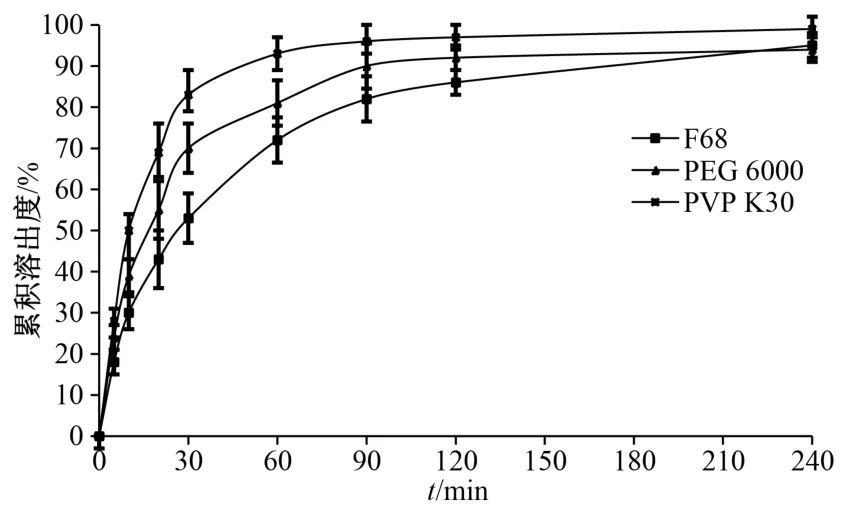
圖1 不同載體下固體分散體溶出曲線Fig.1 Dissolution curves for solid dispersions loaded by different carriers
2.4.2 藥載比 考察藥載比1∶5、1∶6、1∶8、1∶10 時對二氫楊梅素溶出速率、溶出度的影響,結果見圖2。由此可知,藥載比為1∶8 時溶出速率、溶出度均接近1∶10,故選擇其作為藥載比。
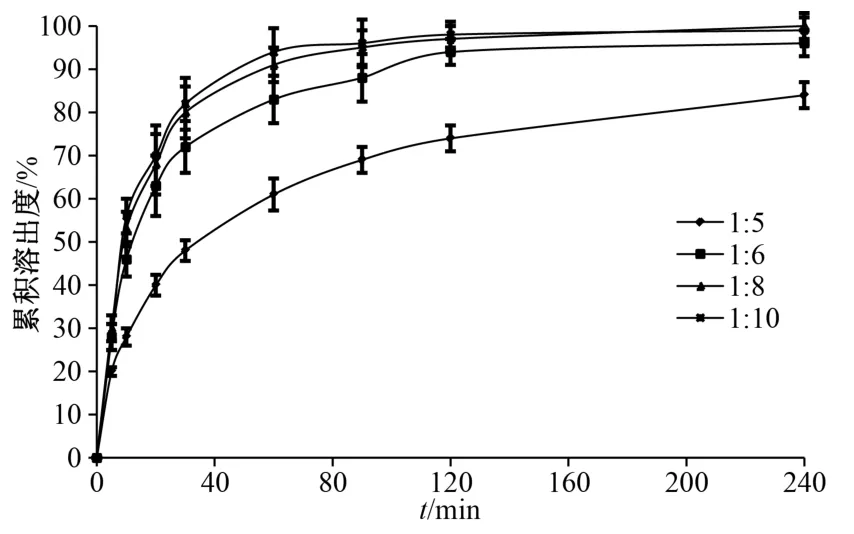
圖2 不同藥載比下固體分散體溶出曲線Fig.2 Dissolution curves for solid dispersions with different drug-carrier ratios
2.5 晶型分析 將二氫楊梅素與PVP K30 按1∶8比例簡單混合,作為物理混合物。掃描條件為銅靶,掃描速度5°/min,掃描范圍3°~45°,管壓40 kV,管流200 mA,結果見圖3。由此可知,二氫楊梅素制備成固體分散體后由結晶型態轉變為典型的無定型態;物理混合物XRD 圖譜中仍可觀察到二氫楊梅素結晶峰,表明簡單的混合并不能改變該成分存在狀態。
2.6 表觀溶解度測定 取過量二氫楊梅素、物理混合物、固體分散體分別加到蒸餾水中,25 ℃下恒溫搖床法振蕩平衡48 h,0.45 μm 微孔濾膜過濾,測定表觀溶解度,結果見表1。由此可知,制成固體分散體后二氫楊梅素表觀溶解度提高了9.8倍,而物理混合物僅小幅提高。
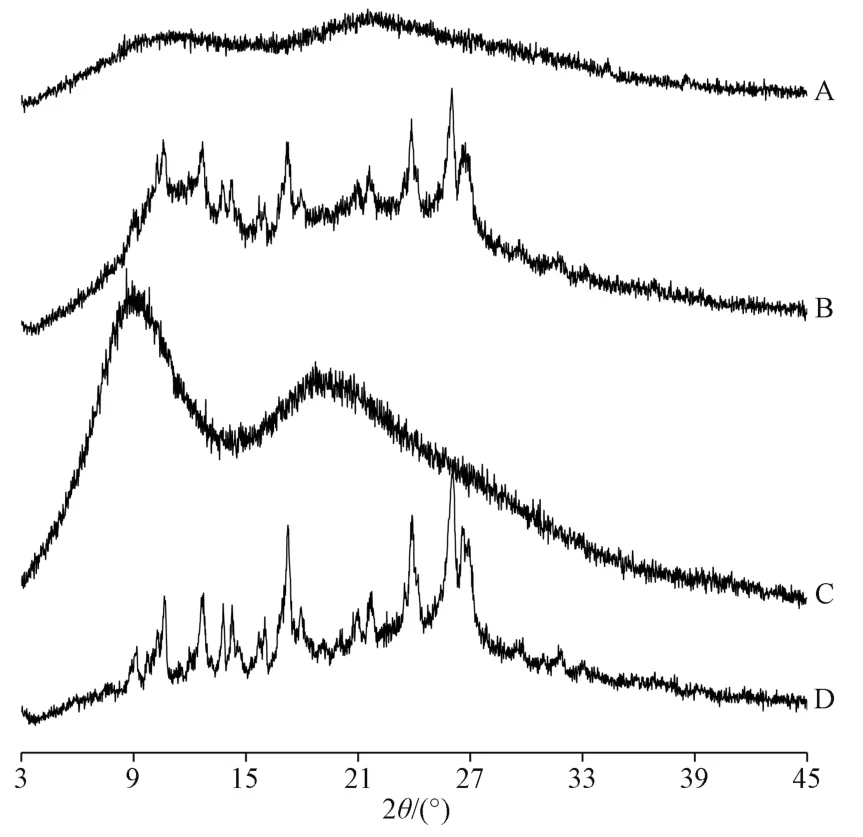
圖3 樣品XRD 圖譜Fig.3 XRD patterns for samples
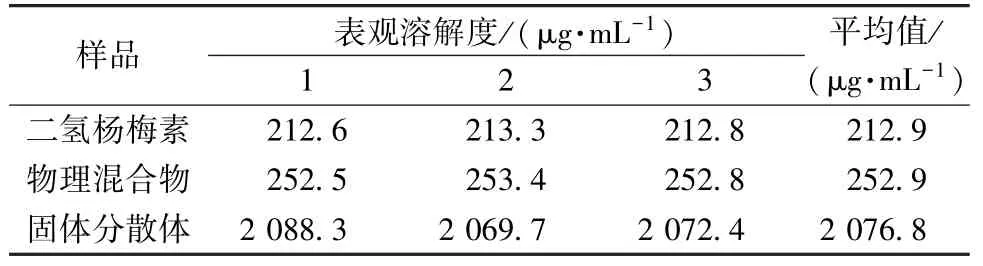
表1 樣品表觀溶解度測定結果(n=3)Tab.1 Results of apparent solubility determination of samples(n=3)
2.7 體外溶出度測定 按“2.2”項下方法測定二氫楊梅素、固體分散體體外溶出度,結果見圖4。由此可知,固體分散體120 min 累積溶出度即接近100%,而二氫楊梅素240 min 時僅為36.49%。
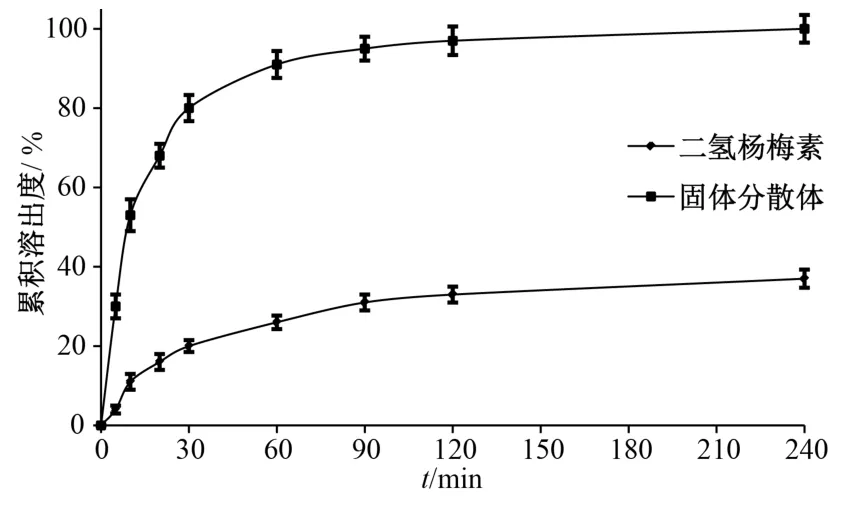
圖4 樣品溶出曲線Fig.4 Dissolution curves for samples
2.8 體內藥動學研究
2.8.1 灌胃液制備 分別取二氫楊梅素、物理混合物、固體分散體適量,0.5% CMC-Na 溶液配制成18.0 mg/mL 混懸液(以二氫楊梅素計),即得。
2.8.2 分組、給藥、采血[14]取SD 大鼠18 只,隨機分為3 組、按150 mg/kg 劑量(以二氫楊梅素計)分別給予“2.8.1”項下3 種灌胃液,于0.167、0.25、0.5、0.75、1、2、4、6、8、12 h玻璃毛細管眼眶取血各約0.3 mL,置于肝素浸潤的離心管中,混勻后迅速離心(4 000 r/min)2 min,轉移上層血漿至另一空白離心管中,冷凍保存。
2.8.3 血漿樣品處理 配制200 mL 提取溶液正己烷-二氯甲烷-異丙醇(100∶60∶10),備用。吸取血漿樣品100 μL、內標溶液50 μL,置于離心管中,渦旋1 min,加入4 mL 提取溶劑再渦旋5 min,高速離心(10 000 r/min)15 min,分取上層有機相,氮氣吹干,殘渣用100 μL 流動相復溶。
2.8.4 方法學考察 精密稱取10.0 mg 芹菜素對照品至10 mL 量瓶中,乙腈超聲溶解,流動相定容,再用流動相稀釋至1.0 μg/mL,作為內標溶液。取“2.1.2”項下不同質量濃度對照品溶液,大鼠空白血漿分別制成0.1、0.5、1.0、2.5、5.0 μg/mL含藥血漿,精密量取100 μL,加入50 μL內標溶液,按“2.8.3”項下方法處理。以溶液質量濃度為橫坐標(X),二氫楊梅素與內標峰面積之比為縱坐標(Y)進行回歸,得方程為Y=0.040 9X-4.025 4(r=0.990 9),在0.1~5.0 μg/mL范圍內線性關系良好。取空白血漿、灌胃給藥12 h 血漿樣品加內標、血漿對照品溶液(0.1 μg/mL)適量,在“2.1.1”項色譜條件下進樣測定,結果見圖5,可知血漿內源性物質不干擾內標、二氫楊梅素色譜峰,該方法專屬性良好。
2.8.5 方法學考察 取低(0.1 μg/mL)、中(2.5 μg/mL)、高(5.0 μg/mL)質量濃度含藥血漿,測得日內、日間精密度RSD 分別小于7.11%、9.94%,表明該方法精密度良好。將測定質量濃度與實際配制質量濃度比較,計算回收率,測得在87.58%~92.44%之間。血漿樣品保存于-20 ℃冰箱中,于0、1、2、3、4、5 d 測定二氫楊梅素與內標峰面積的比值,測得RSD 為7.90%;置于室溫下,于0、1、2、6、8、12 h 測定,測得RSD 為6.72%,表明樣品在放置、測定過程中穩定性良好。取含藥血漿進行逐步稀釋,以信噪比S/N=10為定量限,S/N=3 為檢測限,測得兩者分別為0.02、0.005 μg/mL。綜上所述,上述方法可用于二氫楊梅素血藥濃度測定。
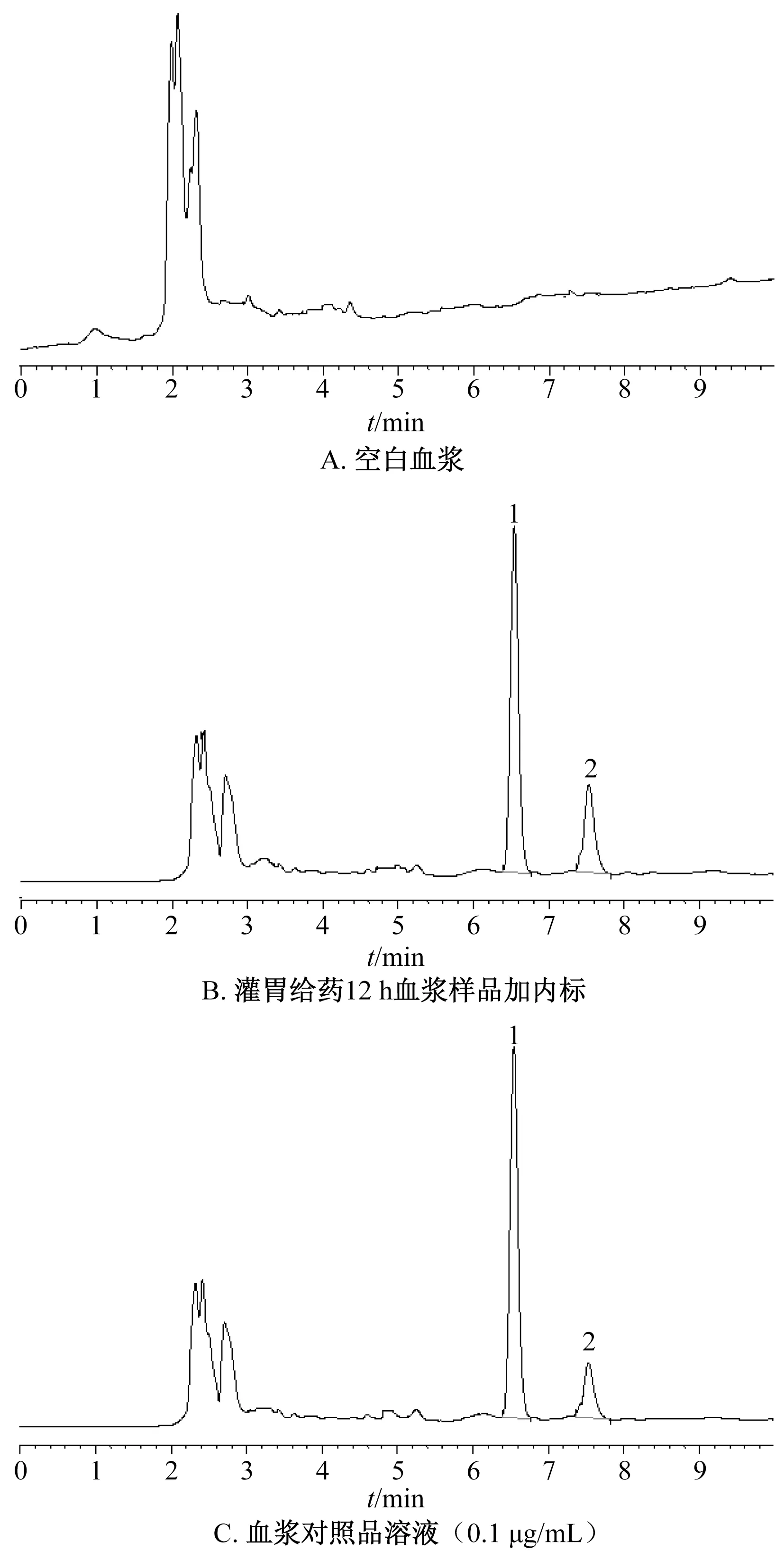
圖5 二氫楊梅素HPLC 色譜圖Fig.5 HPLC chromatograms of dihydromyricetin
2.8.6 分析結果 主要藥動學參數采用3P97 程序統計矩模型計算,AUC、Cmax經對數轉換后采用獨立樣本t檢驗,t1/2、tmax采用非參數法秩和檢驗,結果見圖6、表2。由此可知,與原料藥、物理混合物比較,固體分散體tmax縮短(P<0.05),Cmax、AUC0~t、AUC0~∞升高(P<0.05,P<0.01),相對生物利用度提高了2.66 倍。
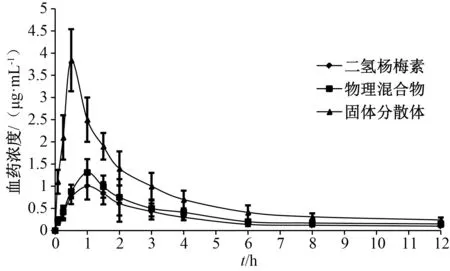
圖6 樣品血藥濃度-時間曲線Fig.6 Plasma concentration-time curves for samples
表2 樣品主要藥動學參數(±s, n=6)Tab.2 Main pharmacokinetic parameters for samples(, n=6)
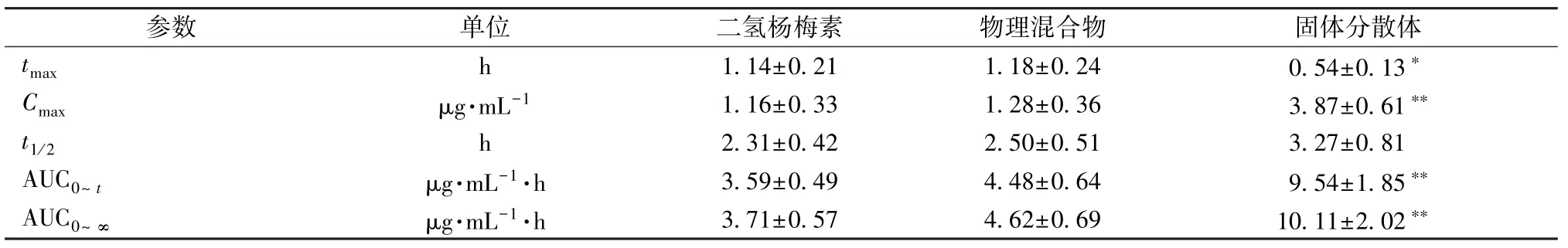
表2 樣品主要藥動學參數(±s, n=6)Tab.2 Main pharmacokinetic parameters for samples(, n=6)
注:與二氫楊梅素或物理混合物比較,?P<0.05,??P<0.01。
3 討論
二氫楊梅素溶解度僅為212.9 μg/mL,在240 min內累積溶出度僅為36.49%,而將該成分制成固體分散體后,溶解度可提高至9.8 倍,體外溶出速率、累積溶出度均得到明顯改善,為促進藥物吸收、提高生物利用度奠定了基礎。研究表明,固體分散體提高溶解度、溶出速率可能與減小顆粒粒徑(以分子或微晶狀態存在于固體分散體中)、增強潤濕性(親水性高分子材料)、提高藥物疏松程度、改變藥物晶型(轉變為無定型狀態)等作用有關[14-16]。
二氫楊梅素脂質體、自微乳、微囊等[8-10]制劑技術制備工藝復雜,其中制備自微乳需使用大量表面活性劑,而二氫楊梅素脂質體或微囊包封率均不高,分別僅為54.7%[8]、66.13%[10],可能與該成分本身理化性質有關[17-18]。蔣才武等[19]曾以PEG 6000 為載體,對二氫楊梅素固體分散體進行了研究,而本研究發現采用PVP K30 材料時,固體分散體溶出速率和藥物累積溶出度均高于PEG 6000,并且當藥物、載體材料用量比例1∶8 時,120 min內二氫楊梅素基本溶出完畢;與原料藥相比,固體分散體tmax明顯提前,可能與該技術可加快藥物溶出、提高溶出速率等有關[12],同時Cmax、相對生物利用度明顯提高,可能是由于該技術可提高藥物溶解度、溶出度,促進藥物吸收。
藥物在體內順利被吸收除了與其本身溶解度有關外,還與其透膜能力(脂溶性)相關,文獻[15,20]報道,固體分散體技術雖然能極大改善難溶性藥物水溶性、溶出度,但對其脂溶性影響很小。此外,藥物吸收還可能受到胃腸道環境等因素影響[13],從而使生物利用度提高程度受到一定限制。本實驗成功制備了二氫楊梅素固體分散體,其工藝簡單,材料安全,促進藥物吸收結果明顯,今后將對其藥效學進行評價[21],以期為相關制劑研發提供更全面可靠的依據。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
云南醫藥(2019年3期)2019-07-25 07:25:14
現代檢驗醫學雜志(2016年5期)2016-08-20 03:16:56
海南醫學(2016年8期)2016-06-08 05:43:00
西南軍醫(2016年5期)2016-01-23 02:20:33
川北醫學院學報(2015年5期)2015-12-05 08:22:28
醫學研究雜志(2015年9期)2015-07-01 17:28:15
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
