CD14/TLR1-TLR2/p38 MAPK信號通路在萊姆病發病機制中的作用
2020-04-15 08:36:00陸麗紅簡苗苗計震華寶福凱柳愛華
生命科學研究 2020年1期
關鍵詞:信號
陸麗紅,丁 喆,簡苗苗,計震華,寶福凱*,柳愛華*
(昆明醫科大學a.第二臨床學院; b.云南省高校熱帶傳染病重點實驗室; c.熱帶醫學研究所; d.病原生物學與免疫學系;e.生物化學與分子生物學系,中國云南 昆明 650500)
萊姆病(Lyme disease,LD)又稱萊姆包柔體病(Lyme borreliosis),是由蜱傳播的一種自然免疫源性感染性疾病,伯氏疏螺旋體(Borrelia burgdorferi,Bb)為致病源[1~2]。我國于 1985年首次在黑龍江省林區發現本病病例,現分布范圍已遍及中國大部分地區,至少累及30 個省份[3]。萊姆病系全身性感染,可導致多系統多臟器損傷[4],臨床表現較復雜,一般潛伏期為3~32 d,平均7 d,臨床癥狀分為一、二、三期,一期表現為皮膚慢性游走性紅斑,可伴有乏力、畏寒發熱、惡心嘔吐和腦膜刺激征等臨床癥狀; 二期時8%~15%的患者出現神經系統癥狀和心臟受累的現象; 三期時80%的患者出現不同程度的關節癥狀[5]。萊姆病的致死率和致殘率逐年上升,但其發病機制仍不清晰。近年來,人們在探索CD14/TLR1-TLR2/p38 MAPK 信號通路各部分介導伯氏疏螺旋體引發的炎癥反應、內化及宿主細胞凋亡方面取得了很大進展,為萊姆病的發病機制研究提供了重要啟示。
1 CD14/TLR1-TLR2/p38 MAPK信號通路概述
伯氏疏螺旋體入侵機體后,CD14 識別病原體表面的細菌脂蛋白(bacterial lipoprotein,BLP),二者形成穩定的復合物[6]。該復合物與Toll 樣受體2 (Toll-like receptor 2,TLR2)結合后引起 TLR1 和TLR2 的二聚化,同時可引起二聚體的構像變化,使得TLR1 和TLR2 的TIR 結構域更加靠近[7]。TLR1和TLR2 通過脂蛋白N-端的半胱氨酸識別三酰化的脂蛋白[8]。TLR1-TLR2 的配體激活髓樣初級反應基因88 (myeloid differentiation primary response gene 88,MyD88)依賴通路[9],其中TLR2 通過BB-loop 和poc 位點募集下游接頭分子MyD88,完成TLR2 的TIR 結構域與MyD88 的TIR 結構域的二聚化[10]。但有研究表明TLR1-TLR2 除激活MyD88外,也激活了TRIF(TIR-domain-containing adaptor inducing interferon-β),激活的 TRIF 可介導TLR1-TLR2 信號來合成配體,如 Pam3CSK4[11]。TRIF 和MyD88 相互作用,引起下游信號轉導,其中Petnicki-Ocwieja 等[11]證明TRIF 誘導促炎因子需要MyD88 的存在,但其相關機制尚不明確。在巨噬細胞中CD14、TLR1-TLR2 和吞噬細胞受體形成脂筏[12],導致原本結合在MyD88 調節蛋白Toll 互作蛋白(Toll-interacting protein,Tollip)上的MyD88的下游激酶——白介素1 受體相關激酶(interleukin-1 receptor-associated kinase,IRAK)從結合體上脫落,同時完成IRAK 的自動磷酸化[13]。MyD88的 C 端 Toll/IL-1 受體(Toll/IL-1 receptor,TIR)結構域與受體結合,N 端的DD 結構域募集IRAK1 和IRAK4,募集的IRAK4 發揮激酶作用導致IRAK1磷酸化[14]。活化后的IRAK1 進一步募集腫瘤壞死因子受體相關因子6(tumour necrosis factor-receptorassociated factor-6,TRAF6)并使其寡聚化。IRAK1與TRAF6 隨后與轉化生長因子β 活化激酶 1(transforming growth factor beta-activated kinase-1,TAK1)、TAK1 結合蛋白 1(TAK1-binding protein-1,TAB1)和TAB2 形成一個復合體。復合體包含了泛素結合酶13(ubiquitin-conjugating enzyme-13,Ub-C13)和泛素結合酶E2 變體-1A (ubiquitin-conjugating enzyme E2-variant-1A,UEV1A)[15],介導了TRAF6 的泛素化。泛素化的TRAF6 通過與Toll途徑進化保守信號中介分子(evolutionarily conserved signaling intermediate,ECSIT)相互作用激活TAK1。TAK1 通過激活絲裂原活化蛋白激酶激酶3 (mitogen-activated protein kinase kinase-3,MKK3)和MKK6 使p38 磷酸化。磷酸化的p38 促進吞噬體的形成,激活下游激酶,引起細胞核因子κB (nu clear factor-κB,NF-κB)的核易位,穩定促炎細胞因子的mRNA,使促炎因子表達增加[12](圖1)。
2 CD14/TLR1-TLR2/p38 MAPK信號通路與萊姆病相關炎癥
由于缺乏脂多糖(lipopolysaccharide,LPS),細菌脂蛋白(BLP)被認為是介導伯氏疏螺旋體的主要炎癥反應介質[16]。伯氏疏螺旋體的脂蛋白是一種膜錨定蛋白,由脂蛋白前體經脂質化修飾而成,并經由Lol (localization of lipoproteins)系統從細胞周質運輸到細胞外膜上[8]。BLP 被認為是炎癥的主要誘導因子,作為TLR2 配體的同時也是一種病原相關分子模式(pathogen-associated molecular pattern,PAMP),并且在極低的濃度下也能被TLR2識別。CD14 可識別伯氏疏螺旋體表面的脂蛋白,從而激活CD14/TLR1-TLR2/p38 MAPK 通路,誘導促炎因子分泌,引起機體的炎癥反應。
2.1 CD14與萊姆病相關炎癥
CD14 的編碼基因位于人的第5 號染色體長臂端5q23~q31,約含有1 338 個核苷酸殘基,編碼含有375 個氨基酸的糖蛋白,該糖蛋白的相對分子質量為55 kD,主要特征結構為N-末端富含亮氨酸的重復單位(leucine-rich repeats,LRRs)[17]。CD14 分為 mCD14 和 sCD14,mCD14 主要分布在單核細胞、巨噬細胞和樹突狀細胞的表面,sCD14則存在于正常人和動物的血漿中,二者的區別在于 sCD14 較 mCD14 缺少 GPI 螯合物[18]。CD14 作為一種連接受體[19],主要的生物學功能是識別、結合LPS 或LPS/LBP 復合物,介導LPS 所致的細胞反應,其在炎癥反應、內毒素休克等病理反應中起重要作用。在萊姆病中,CD14 介導的MAPK 通路在促炎抗炎微環境的調控中扮演了重要角色,參與了螺旋體刺激巨噬細胞后白介素10 (interleukin-10,IL-10)和腫瘤壞死因子α (tumor necrosis factor-α,TNF-α)的生成[20]。CD14 可在多種損傷相關分子模式(damage-associated molecular patterns,DAMPs)作用下促進炎性細胞分泌促炎因子。研究報道,缺少CD14 的細胞在DAMPs 的作用下表現出低反應的特性,下游信號如p38 MAPK 等均不能被激活,導致病原體入侵機體時機體分泌更多的促炎因子,從而表現出更嚴重的炎癥反應[12]。此外,在萊姆關節炎中,CD14 的缺乏會導致基質金屬蛋白酶9 的活化減少,膠原蛋白降解減少,殺滅螺旋體的中性粒細胞募集受阻,最終使萊姆關節炎加重[21]。由此可見,CD14 在機體炎癥反應中發揮著重要作用。
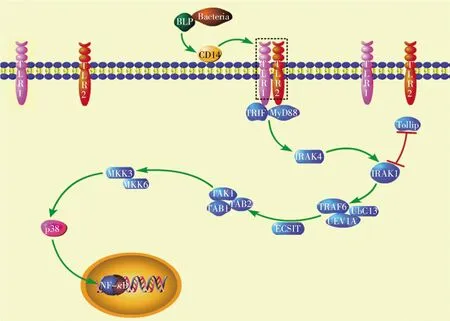
圖1 細菌脂蛋白介導的CD14/TLR1-TLR2/p38 MAPK 信號通路Fig.1 CD14/TLR1-TLR2/p38 MAPK signaling pathway mediated by bacterial lipoproteins
2.2 TLR1/TLR2與萊姆病相關炎癥
TLRs 是表達于天然免疫細胞、內皮細胞和上皮細胞等細胞表面的重要模式識別受體,通過自身的LRRs 結構域識別PAMPs,刺激炎癥因子的生成,參與病原體的殺傷[10]。TLR1 和TLR2 分布于細胞膜表面,參與識別病原體表面的脂蛋白、鞭毛和脂肽等成分。和TLRs 家族其他成員相同,TLR1 和TLR2 含有胞內保守的TIR 結構域,用于與下游接頭分子的TIR 功能域特異性相互作用。但是,TLR2 的激活常需要TLR1 和TLR6 的參與,且TLR2 需與TLR1 和TLR6 分別結合形成異二聚體[22]。TLR1 中保留了與TLR2 結合的二聚體界面和脂肽通道,TLR2 和TLR1 以αE 螺旋的FW殘基為關鍵位點[10],通過螺線管結合形成異質二聚體[22]。除 αE 螺旋的 FW 殘基外,BB-loop 和 DD-loop 也是TLR1 和TLR2 異源二聚化的重要功能域[10]。TLR2 之所以能夠識別細菌、真菌和原生生物等不同微生物的外膜成分,TLR1 在其中扮演著重要角色。TLR1 具有一個疏水性通道,對于TLR1 和TLR2 形成異二聚體識別三酰化的脂蛋白有著重要作用。相關結構研究表明,三酰化的脂肽類似于革蘭氏陰性桿菌的脂蛋白,且TLR1/TLR2 一般只能夠識別三酰化的脂蛋白[7,23]。伯氏疏螺旋體表面的脂蛋白主要由TLR1-TLR2 識別[24],識別后可引起下游炎癥信號的激活,并使機體產生促炎因子。在樹突狀細胞中,TLR1-TLR2 識別伯氏疏螺旋體后激活MyD88 依賴的NF-κB 信號級聯反應,導致樹突狀細胞的成熟和遷移; 樹突狀細胞作為一種專職抗原提呈細胞(APC),將伯氏疏螺旋體提呈給CD4+T 細胞,促進促炎因子生成[25]。相關研究報道,在敲除了TLR2 的小鼠萊姆關節炎模型中CD4+T 細胞并未發生增殖,而CD8+T細胞發生了增殖; 同時,CD8+T 細胞可以刺激滑膜細胞產生CXCL9,加速炎癥進程[24]。另有研究報道,在敲除IL-10 的小鼠中,TLR2 可以激活CD4+T細胞和 CD8+T 細胞,促進干擾素-γ (interferon-γ,IFN-γ)的產生,而伯氏疏螺旋體的入侵也可促進CD4+T 細胞和CD8+T 細胞的增殖,并在感染3周后細胞數量達到峰值。此外,在伯氏疏螺旋體感染后,TLR2的轉錄特異性增強,表達量增加,而TLR1、TLR6 等的轉錄特異性不變[26]。綜上所述,CD4+T 細胞和CD8+T 細胞在萊姆關節炎發生中發揮重要作用,而TLR2 則作為T 細胞活化的關鍵成分參與到炎癥的進程中。當用特異性中和抗體阻斷TLR1-TLR2 信號通路時,趨化因子CC 和 CXC 家族的分泌受到抑制,這提示可以通過阻斷TLR1-TLR2 的激活來抑制機體的炎癥反應[27]。
2.3 p38 MAPK與萊姆病相關炎癥
p38 作為MAPKs 家族的三大分支之一,通過微調抗炎和促炎信號影響炎癥的發生,并且在應激、凋亡、細胞周期和生長等多種生理和病理過程中起重要作用[28]。p38 是由360 個氨基酸組成的,大小為38 kD 的蛋白質,與JNK 同屬應激激活的蛋白激酶。目前存在6 種p38 MAPK 亞型,分別是 p38α1/α2、p38β1/β2、p38γ 和 p38δ,其中 p38α和p38β 幾乎在所有的組織細胞中表達[29],但p38γ主要存在于骨骼肌中,p38δ 則多見于睪丸、唾液腺、胰腺、前列腺、小腸、腦垂體等部位,雖然不同亞型其氨基酸的組成個數不同,但同源性超過了50%[30]。p38 MAPK 在萊姆病的發生發展中扮演著重要角色,通過抑制p38 MAPK 可以顯著抑制伯氏疏螺旋體與機體作用時促炎因子的生成,從而減輕萊姆病的嚴重程度。MKK3 是p38 MAPK 的上游激活物,當它被阻斷時,p38 MAPK 不能被激活,Th1 細胞反應降低,經p38 MAPK 誘導產生的IFN-γ 減少,萊姆關節炎減輕[31]。然而,當在萊姆心臟炎中阻斷MKK3 時,炎癥減輕的效果卻不如萊姆關節炎中顯著,這可能與p38 MAPK 的各種亞型在組織中的分布不均勻,且MKK6 也可以激活p38 MAPK 有關[32]。此外,在神經萊姆病中,p38 MAPK 的受阻也可以抑制少突膠質細胞和巨噬細胞生成促炎因子TNF-α[33]。綜上可知,p38 MAPK在萊姆關節炎、萊姆心臟炎以及萊姆神經炎中都起著重要作用。
3 CD14/TLR1-TLR2/p38 MAPK信號通路與伯氏疏螺旋體內化和宿主細胞凋亡
CD14/TLR1-TLR2/p38 MAPK 信號通路在介導伯氏疏螺旋體引發的炎癥反應方面具有重要作用。同樣,此通路上的重要組成成分在伯氏疏螺旋體介導的細胞凋亡和內化過程中也具有重要作用。
伯氏疏螺旋體進入人體后可以引起小膠質細胞和少突膠質細胞的凋亡,導致炎癥的發生,而該炎癥的發生又可引起更多神經元死亡,使機體表現出神經系統的病變; 相關研究認為小膠質細胞在炎癥微環境中對神經元的凋亡發揮著不可替代的作用,當受到伯氏疏螺旋體刺激后,小膠質細胞上的TLR2 表達上調,可能通過增加IL-6 的產生來介導細胞凋亡[34~37]。p38 激酶是一種應激激活的激酶,參與調節炎癥介質的產生[38],并可通過下游的caspase 來誘導細胞凋亡[30]。在關于伯氏疏螺旋體導致細胞凋亡的研究中,相關報道指出TLR2 也可通過MyD88、Fas 相關死亡結構域蛋白(Fas-associated protein with death domain,FADD)/caspase-8 介導與細胞凋亡相關的信號轉導[39]。
細胞的內化也被認為是導致萊姆病的一大重要因素。伯氏疏螺旋體的內化經由固有免疫系統細胞的吞噬作用介導[40]。目前認為伯氏疏螺旋體可通過3 種途徑進入吞噬細胞,分別是Fcγ 受體介導的吞噬作用、傳統的吞噬作用和卷曲吞噬作用[41],其中卷曲作用被認為是伯氏疏螺旋體內化的首選途徑[42]。巨噬細胞可以伸出一個長而富含肌動蛋白的絲狀偽足包圍住伯氏疏螺旋體,且卷曲的偽足有多個彎曲節點,可以靈活地與螺旋體結合,在此過程中細胞肌動蛋白骨架發生重組[43]。當病原體與吞噬細胞表面結合后,肌動蛋白相關蛋白質募集到脂筏表面,進行受體依賴的細胞骨架重排,以此作為伯氏疏螺旋體內化的前奏[12]。許多受體都被證明參與了伯氏疏螺旋體的內化過程,如Fcγ 受體、甘露糖受體、補體受體 3、整合素 αMβ2等[42]。當伯氏疏螺旋體與吞噬細胞結合時,與吞噬有關的基因CD14 表達上調,隨后CD14 與整合素的C 型凝集素結構域結合,促進補體受體3 與伯氏疏螺旋體的結合物向細胞表面移動,從而啟動內化信號[42,44~45]。整合素是配體內吞的重要調節因子,伯氏疏螺旋體可表達整合素α3β1,并通過脂蛋白的內吞作用激活TLR1-TLR2,隨后TLR1-TLR2 的配體Pam3CSK4誘導內吞信號轉導,并通過網格蛋白完成內化。與整合素α3β1 不同,整合素 αvβ3 介導 Pam3CSK4與巨噬細胞結合,導致三酰化脂蛋白與TLR1-TLR2 在細胞表面聚集,從而促進細胞內吞信號轉導[19]。需要指出的是,伯氏疏螺旋體進入宿主細胞并不完全依賴某一個受體,有學者提出在這些受體激活的下游吞噬通路中可能存在著復雜的交叉作用[42]。
4 結語與展望
綜上所述,CD14/TLR1-TLR2/p38 MAPK 信號通路在萊姆病的發病機制中可能扮演著重要角色。已有研究顯示,加入外源性cAMP 能顯著增加IL-10 的表達且能顯著降低促炎、關節炎易感性基因的轉錄水平[20]。因此,cAMP 調節劑相關的藥物研究或許可以為治療伯氏疏螺旋體引起的炎癥反應打開新思路。此外,蛋白激酶C (protein kinase C,PKC)的新型抑制劑粗糠柴毒素和白屈菜赤堿可以顯著抑制p38 的磷酸化,同時可以顯著減輕感染后C3H/HeN 小鼠的踝關節炎和腫脹,并且可以阻斷IL-1b 和IL-8 等炎癥介質的轉錄[46],提示含有PKC 抑制劑的藥物對伯氏疏螺旋體引起的感染可能具有臨床應用價值。
目前,CD14/TLR1-TLR2/p38 MAPK 信號通路與萊姆病相關螺旋體內化、炎癥反應和細胞凋亡的關系都有研究,但存在對三者之間的聯系研究不夠深入的情況。對三者之間的聯系進行深入研究將讓我們對萊姆病的發病機制有更全面的了解。同時,現有工作注重該信號通路上某一成分的研究,缺少對信號通路的整體研究。完整信號通路的研究將為探索萊姆病發病機制提供新思路,并可為一些靶點藥物的開發提供更好的理論依據。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
