10 年隨訪淋巴瘤樣丘疹病1 例
2020-04-30 02:01:08路永紅
皮膚病與性病 2020年2期
陽 眉,李 芬,路永紅
(1.成都市第二人民醫院皮膚科,四川 成都 610017;2.成都市第二人民醫院病理科,四川 成都 610017)
1 臨床資料
患者,男,47 歲,反復軀干及雙下肢紅斑、斑塊、結節、潰瘍10 年,于2016 年6 月29 日來我院就診。10 年前,患者無明顯誘因出現下肢紅斑、結節,瘙癢不明顯,無疼痛。2007 年出現紅斑,且紅斑逐漸擴大變硬,形成斑塊,能自行消退,但反復發生。結節斑塊可破潰結痂,愈合形成大小不一萎縮性瘢痕。患者無乏力、發熱、關節疼痛、消瘦及淋巴結腫大。2007年患者曾在“西南醫院”皮膚科經皮損活檢及免疫組化確診為“淋巴瘤樣丘疹病”,后每6 個月隨訪1次,偶有新發皮損時,曾使用鹽酸米諾環素膠囊0.1,每晚一次,重組人干擾素α-1b 注射液50μg,每周肌注1 次治療。10 年來無明顯自覺癥狀。個人史無特殊,家族中無類似疾病患者。
系統查體:全身淺表淋巴結無腫大及壓痛。專科查體:軀干和四肢可見數十個拇指大斑塊,部分表面糜爛,無壓痛,大量陳舊性萎縮性瘢痕(圖1 ~圖3)。口腔和生殖器未累及。

圖1 ~圖3 軀干和四肢可見數十個拇指大斑塊,部分表面糜爛,無壓痛,大量陳舊性萎縮性瘢痕
2016 年右大腿皮損組織病理:潰瘍邊緣鱗狀上皮瘤樣增生,表皮內中性粒細胞浸潤。真皮內散在或簇狀分布大小不等的異型細胞,部分異型細胞呈R-S 樣。周圍伴淋巴細胞、中性粒細胞、嗜酸性粒細胞浸潤。皮下組織未見確切異型細胞,(圖4 ~圖6)。
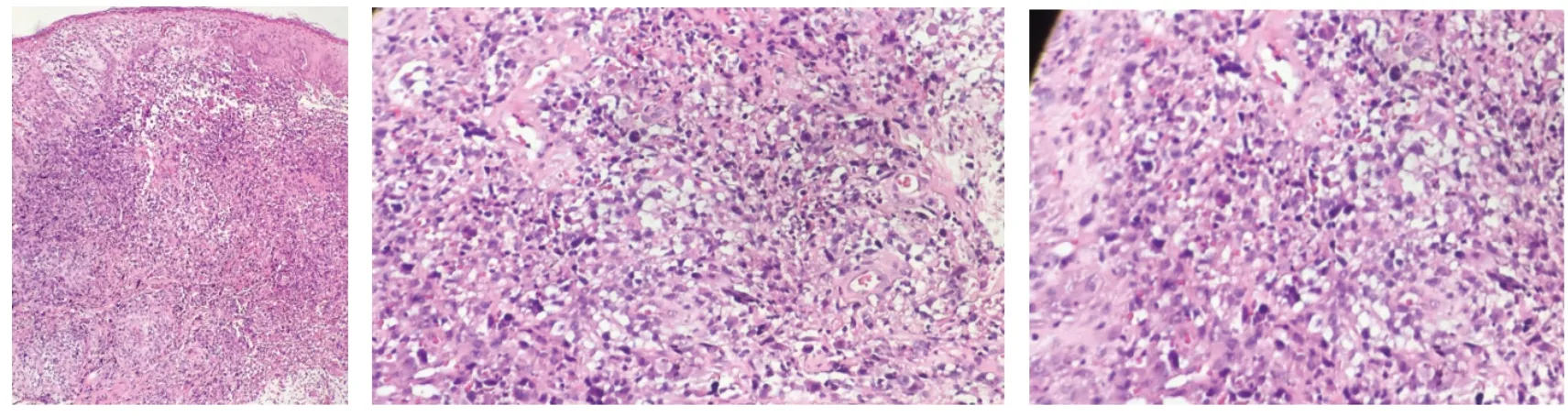
圖4 ~圖6 HE:表皮增生、表皮與真皮分離,真皮內中性粒細胞、異型淋巴細胞、嗜酸性粒細胞及小淋巴細胞浸潤(圖4 ×100,圖5 ~圖6 ×400)
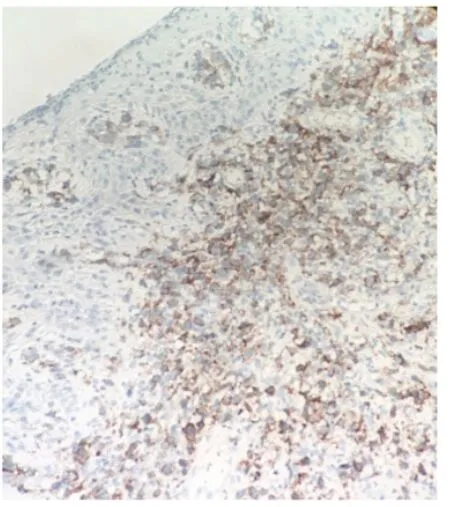
圖7 免疫組化:CD30陽性細胞(棕色信號)散在或簇狀分布(En-Vision 法×200)
免疫組化:CD30(+,陽性細胞呈簇狀或散在小片狀分部)、CD3(個別+,背景中部分小淋巴細胞+)、GranzymeB(+)、TIA-1(+);CD20(-)、CD56(-)、ALK(-)、EMA(-)、S100(-)、HMB45(-)、PCK(-);Ki67 陽性率約80%,(圖7)。
TCRG 基因重排檢測:在目標片段范圍內查見克隆性擴增。結合臨床表現、組織病理和免疫組化診斷:淋巴瘤樣丘疹病(A 型),基因重排檢測提示腫瘤細胞有克隆性增生,建議長期隨訪復查。
本次治療予以“咪喹莫特乳膏、夫西地酸軟膏”外用。電話隨訪,患者病情穩定,無復發。
2 討論
淋巴瘤樣丘疹病(Lymphomatoid papulosis)是一種慢性復發性、自愈性、丘疹壞死性或丘疹結節性皮膚病,臨床上類似急性痘瘡樣苔癬樣糠疹,組織病理表現為CD30陽性的低度惡性淋巴瘤[1]。皮損好發于軀干和四肢,臨床表現為紅棕色丘疹結節,中央可發生出血、壞死和結痂,(3 ~8)周皮損可自然消退,消退后遺留色素斑或色素沉著斑,偶爾遺留淺表萎縮性瘢痕。病程長短不一,可數月甚至40 余年[1]。診斷需要結合臨床、病理、免疫表型,以及必要的長期隨訪。本病有自限性,預后良好,5 年生存率100%,但有5%~20%患者可發展為其他類型淋巴瘤。不同治療對病程轉歸無明確意義,故大多數患者無需特殊治療,但長期的定期隨訪十分重要[2]。
本例患者病史10 年余,軀干、四肢皮損反復發作,可自行消退,臨床表現和病情發展過程較典型,結合組織病理學特征和免疫組化檢查,診斷淋巴瘤樣丘疹病(A 型),在長達10 余年的隨訪觀察中,病情穩定,仍宜長期隨訪。
