SCN9A基因突變癲癇患兒基因型與表型分析
2020-05-08 08:18:54丁健張靜雯郭予雄王林淦張宇昕陳志紅翟瓊香
中國醫學創新
2020年10期
丁健 張靜雯 郭予雄 王林淦 張宇昕 陳志紅 翟瓊香
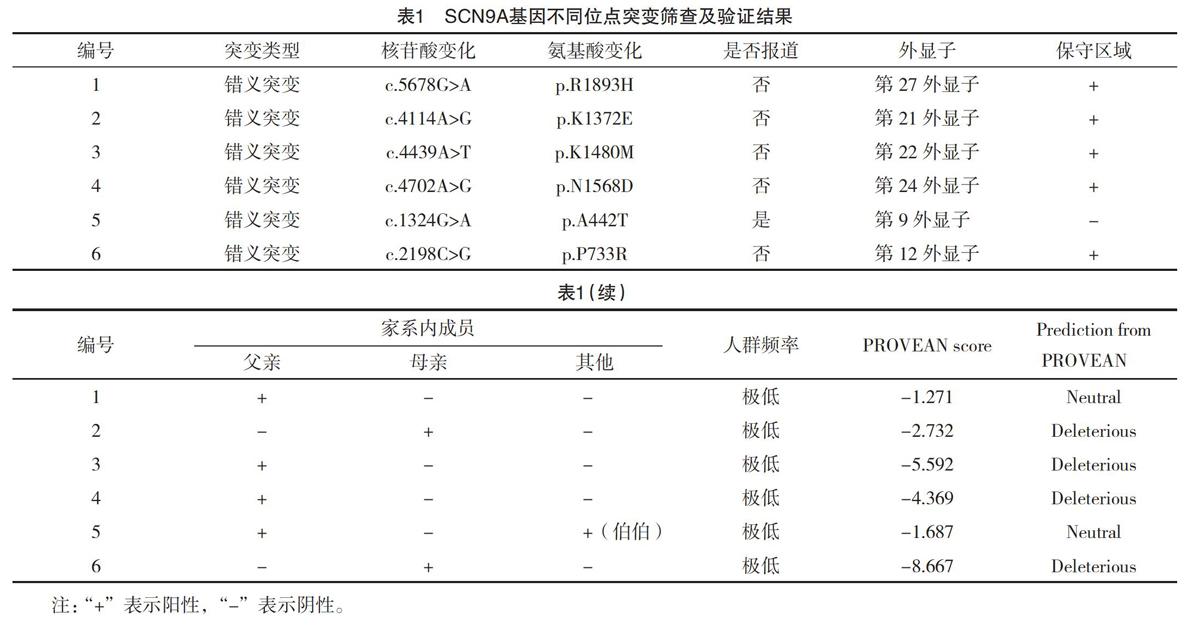
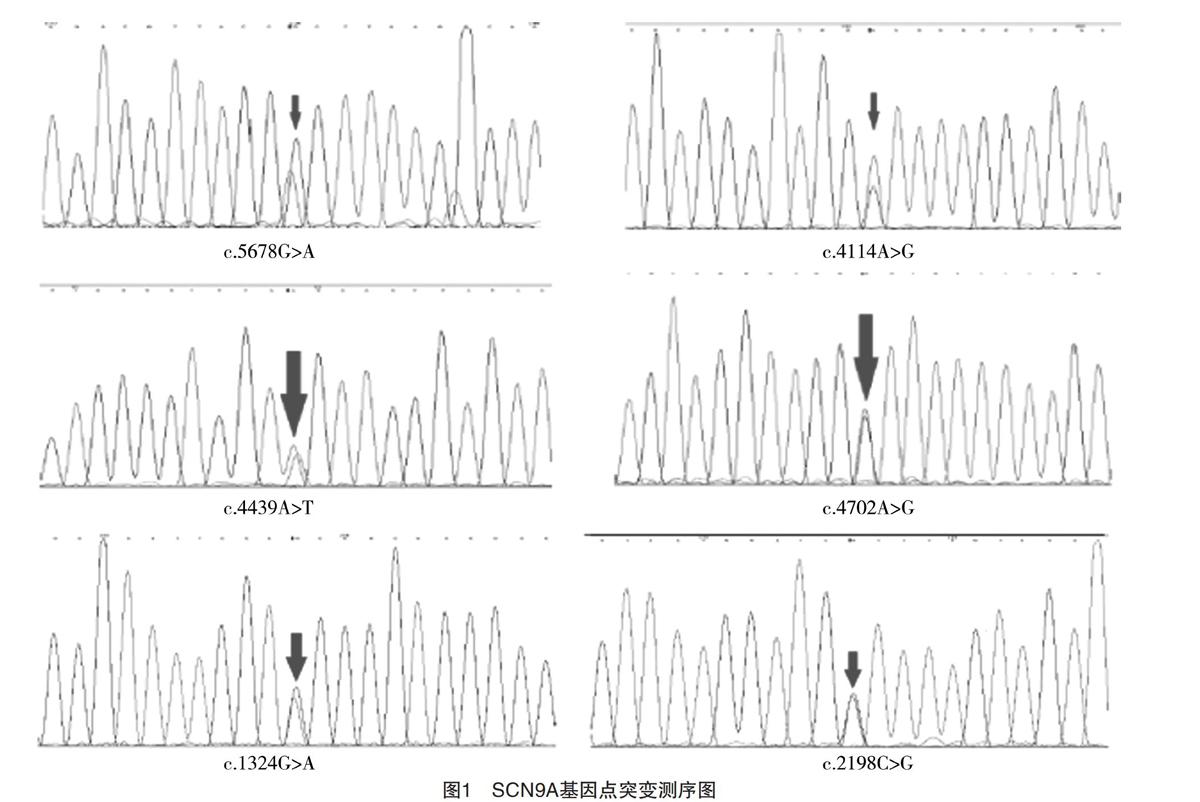

【摘要】 目的:探討SCN9A基因突變癲癇患兒基因型與表型的特點。方法:收集2016年6月-2018年5月廣東省人民醫院兒科就診的癲癇患兒,采用全外顯子組測序篩選突變,并采用Sanger測序方法驗證突變。結果:共發現6例患兒存在SCN9A基因突變,男女各3例,檢測到6個不同位點的突變,均為雜合錯義突變,且為遺傳性突變,導致了氨基酸改變,其中5例突變位點分布在高度保守的區域。臨床發作類型:4例(66.7%)全面性強直-陣攣發作,1例(16.7%)局灶性發作,1例(16.7%)失張力發作;3例(50.0%)患兒發作有熱敏感特點。有多種臨床表型,熱性驚厥附加癥1例,遺傳性癲癇伴熱性驚厥附加癥1例,Dravet綜合征1例,額葉癲癇1例,不明分類的癲癇2例。結論:SCN9A基因突變以雜合的錯義突變為主,大部分突變定位在高度保守的區域,發作類型以全面性發作為主,多數與熱相關,臨床表型譜廣。
【關鍵詞】 SCN9A基因 突變 癲癇 表型
Genotype and Phenotype Study of Children with Epilepsy and SCN9A Gene Mutation/DING Jian, ZHANG Jingwen, GUO Yuxiong, WANG Lingan, ZHANG Yuxin, CHEN Zhihong, ZHAI Qiongxiang. //Medical Innovation of China, 2020, 17(10): 0-030
[Abstract] Objective: To explore the characteristics of genotype and phenotype in children with epilepsy and SCN9A gene mutation. Method: Epileptic patients who treated in the Pediatric Department of Guangdong Provincial Peoples Hospital from June 2016 to May 2018 and detected SCN9A mutation by whole-exome sequencing, and mutations were validated using the Sanger sequencing method. Result: A total of 6 patients (3 boys and 3 girls) with SCN9A mutations were collected. Six SCN9A mutations were identified. All of them were heterozygous missense mutation and inherited from one of their parents, caused amino acid changes. Five mutations of them were distributed in highly conservative regions. The generalized tonic-clonic seizure: the most common seizure type was observed in 4 patients (66.7%), focal seizure was observed in 1 patient (16.7%), Atonic seizure was observed in 1 patient (16.7%). In 3 patients (50.0%), seizures manifested fever-sensitive. There were many clinical phenotypes, 1 patient was diagnosed febrile seizures plus, 1 patient with genetic epilepsy with febrile seizures plus, 1 patient with Dravet syndrome, 1 patient with frontal lobe epilepsy, 2 patients with unclassified epilepsy. Conclusion: The features of patients with SCN9A mutations include that heterozygous missense mutations are dominant, and most of these mutations distribute in highly conservative regions, the main seizure type is the generalized seizure, most seizures manifeste fever-sensitive, the clinical phenotype spectrum is broad.
[Key words] SCN9A gene Mutation Epilepsy Phenotype
First-authors address: Dongguan Peoples Hospital, Dongguan 523059, China
doi:10.3969/j.issn.1674-4985.2020.10.007
癲癇是一種常見的、反復發作的中樞神經系統疾病,重復多次的癲癇發作可導致大腦神經元細胞的損傷,影響患者的認知功能、學習能力、生活質量等多方面,嚴重者可以致殘甚至危及生命。引起癲癇發作的原因受諸多因素的影響,例如遺傳因素、大腦結構的異常、代謝紊亂、免疫功能障礙、顱內感染以及一些未能明確原因[1]。癲癇按病因可以分為癥狀性、特發性和隱源性癲癇,特發性癲癇與遺傳有密切關系,據統計大概有40%的癲癇患者是遺傳因素引起的,特別是兒童癲癇患者[2]。……
登錄APP查看全文
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(2015年4期)2015-01-23 01:19:30
中國中醫藥現代遠程教育(2014年20期)2014-03-01 04:31:21
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
