探討PM2.5暴露的肺外毒理效應對絕經后骨代謝的影響機制
2020-06-06 08:40:32劉宗玉黃惠娟楊帆曾小娟
中國骨質疏松雜志 2020年4期
關鍵詞:水平
劉宗玉 黃惠娟 楊帆 曾小娟
廈門大學附屬東方醫院婦產科,福建 福州 350025
絕經后骨質疏松癥(postmenopausal osteoporosis, PMOP)是衰老相關性低雌激素效應,導致高效骨重塑向骨吸收方向進行致骨脆性增加、易于骨折的全身性骨病,是骨質丟失中最常見的一種類型[1]。我國人口基數大,人口老齡化嚴峻,上海部分地區調查發現,骨質疏松造成髖部骨折中,中老年女性是男性3倍多,人均住院費用高達3萬余元,平均住院天數近18 d[2]。骨質疏松的高發病率及其并發癥的高致殘率、高致死率及高消耗醫療、經濟資源問題,使得PMOP成為我國醫療衛生面臨的重大挑戰。PM2.5易吸附大量氧化還原特性的過渡金屬、持久的自由基、多環芳烴(PAHs)等有害物質,并具有極強的穿透力,極易破壞肺泡毛細血管屏障隨血液作用于全身臟器,改變機體多種生物活性,包括炎癥細胞的招募,釋放大量炎性因子和活性氧(ROS)等,對人體毒害最強[3-5]。我國京津冀、長三角,珠三角年平均PM2.5分別為70、72、53μg/m3[6]均高于我國規定的二級年均水平限值35μg/m3,提示我國環境污染嚴峻。環境污染對破骨/成骨細胞的殺傷作用與氧化應激及活性氧等相關,其可使RANK/RANKL/OPG比值失衡和下調抗氧化酶, 引起骨質丟失[7]。現就氧化應激、炎癥因子、雌激素及相關受體(如AhR)等對骨質疏松的影響進行綜述。
1 PM2.5暴露的毒性效應與絕經后骨代謝密切相關
1.1氧化應激及活性氧
PM2.5參與機體生物活性轉化過程,可誘導炎癥和氧化損傷產生具有協同效應的活性氧或氮(如ROS、RNS)和炎癥介質[8]。氧化應激及活性氧被廣泛認為是許多病理條件下的致病因素,包括骨質疏松。缺氧可使骨髓基質細胞中的線粒體產生大量活性氧,其在低雌激素水平能強有力促進骨吸收[9-11]。此外,雌激素缺乏可致相關組織主要抗氧化劑、抗氧化酶水平顯著下降,活性氧(如H2O2)水平升高,降低骨的抗氧化防御能力,運用抗氧化劑可阻止卵巢切除后引起的骨質疏松[11-14]。Altindag等[15]通過測定血漿TAS、TOS水平獲得OSI時,發現血漿中TOS和OSI值在絕經后骨質疏松癥患者中明顯升高,血漿TAS水平明顯降低,而腰椎和股骨頸部位骨密度與OSI呈顯著負相關。Baek等[16]通過測定血清DNA氧化損傷的標志物8-羥基-2O-脫氧鳥苷(8-OH-DG)水平,同樣發現8-OH-DG水平與腰椎、全髖關節、股骨頸和粗隆部的骨密度水平呈負相關。
另外,職業性暴露于低水平PM2.5(12.4±6.9 μg/m3)和BaP(1.0±0.6 ng/m3)的出租車司機血清炎癥生物標志物水平明顯升高,活性氧和相關氧化損傷標志物增加,抗氧化酶和抗炎介質(IL-10)下降[17]。反過來,升高的活性氧又可增加吸收性炎癥因子的表達間接刺激破骨細胞[18-19]。雌激素缺乏致骨髓微環境中負調控炎癥因子、氧化物等系統地和局部地增加,并通過TGF-β的表達延緩破骨細胞的凋亡,誘導破骨細胞分化,引發骨質疏松[20]。促炎細胞因子被證明能提高不同類型細胞內的ROS水平[21]。而外源性ROS或內源性ROS均可刺激破骨細胞的形成和吸收活動,但其細節尚不清楚。PM2.5可促進細胞內活性氧生成和NF-κB磷酸化,ROS(主要H2O2)可刺激M-CSF和RANKL的表達,提高RANKL/OPG的比值,使低雌激素水平婦女骨吸收增加和骨量降低[15-16,22]。最新研究指出,RANKL能參與Bach 1的核易位,抑制Nrf 2依賴的抗氧化酶的表達,并通過與Bach 1核蓄積競爭,從而增強破骨細胞內的ROS信號[23]。應用抗氧化劑能減少RANKL誘導的Akt、NF-kB和ERK活化,顯著降低RANKL誘導的骨吸收活性和破骨細胞存活率,促進破骨發生[22]。此外,ROS還可通過MAPK和Ca2+介導的信號通路參與骨代謝[21]。
1.2炎癥細胞因子
目前國內關于PM2.5誘發促炎因子直接或間接引發絕經后骨質疏松的相關研究并不多見。Zheng等[24]發現絕經后婦女腰椎骨密度與血清炎癥細胞因子IL-1、IL-6和TNF-α呈負相關性,提示雌激素可調節各種細胞因子的分泌或釋放阻止骨質丟失。Terauchi等[25]指出骨髓中Th細胞分泌的IL-1、IL-6和TNF-α等均可促進骨髓破骨細胞的發生、增殖,引發骨質疏松。PM2.5在肺中的沉積可引起全身炎癥反應,并能刺激骨髓產生廣譜的促炎細胞因子,且顆粒直徑越小,血清中產生的炎癥細胞因子越多,引發相應臟器病理損害越嚴重[26]。研究發現慢性暴露于PM2.5可使血清中的TNF-α、IL-1、IL-6、IFN-γ等炎癥生物標記物水平明顯升高,導致血清RANKL值增加41%,OPG下降22%,引起該群體骨量減少[17,27-28]。
炎癥細胞因子在缺氧、感染和炎癥期間對骨破壞發揮著關鍵作用。雌激素缺乏可通過抗原提呈細胞和細胞因子IFN-γ、IL-7和TGF-β介導的復雜細胞機制誘導系統地和局部地增加炎癥細胞因子分泌或釋放[25]。強效骨吸收因子(TNF-α、IL-1、IL-6)能通過分布在破骨細胞前體上相應的受體(如TNFR、IL-1 R和IL-6R)發揮其生物學效應,在對其相應受體缺陷小鼠的分析表明,TNFR 1、IL-1 RI、sIL-6R促進破骨細胞分化,而TNFR 2、IL-1 RII抑制破骨細胞分化,TNF-α、IL-1、IL-6能增強TNFR 1、IL-1 RI、sIL-6R破骨發生,降低TNFR 2、IL-1 RII功能,促進破骨發生[29-31]。研究發現敲除TNF-α、IL-1 R、IL-6基因的動物可抑制卵巢切除后骨質丟失,證實細胞因子對絕經后骨代謝的影響是必不可少的、多途徑的[32-34]。
最后,炎癥介質直接或間接通過激活共同信號通路OPG/RANKL/RANK調節骨代謝。這些細胞因子可提高RANKL mRNA的表達和RANKL/OPG的比值,激活下游信號磷酸化(如JNK、p38、MAPK、JAK2、ERK和Akt)促進成骨細胞介導的破骨細胞分化、成熟;還可直接強烈激活NF-κB、p38、ERK和應激激活蛋白激酶/c-Jun NH2-末端激酶活性,并顯著降低ALP活性及成骨細胞基因(RunX 2、Osterix和骨鈣素)的表達,且呈劑量依賴性抑制骨礦化和成骨細胞分化,從而導致女性骨質丟失加重[34-38]。炎癥因子還能強烈抑制成骨細胞的生成,這種抑制成骨細胞活性的途徑是通過影響細胞因子下游信號通路,諸如MAPK、Stats、SMURF 1/2、PGE2等[36,38-43],使新骨形成無法代償骨吸收。
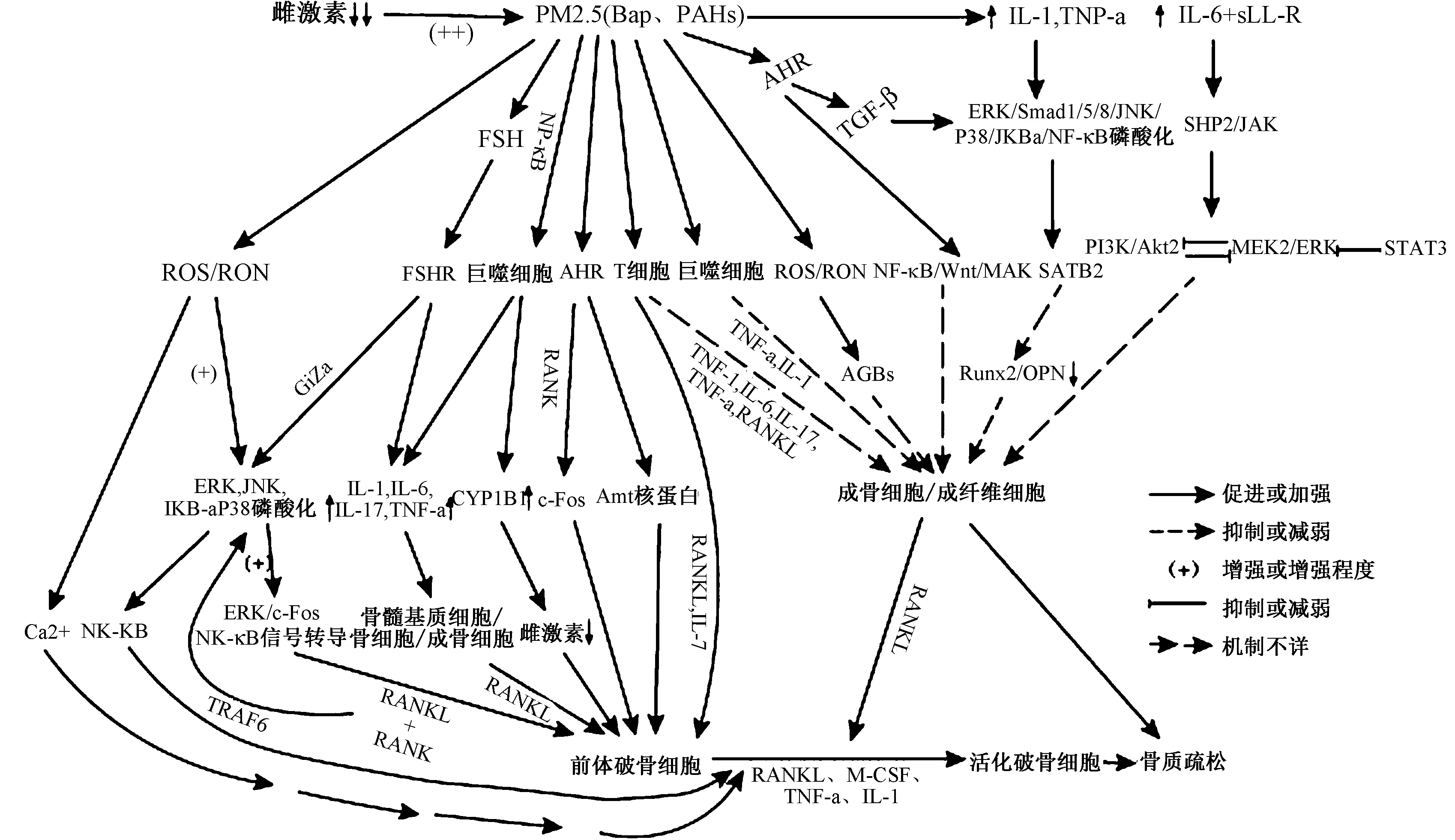
圖1 PM2.5在低雌激素水平中對骨代謝影響的機制(具體機制詳見文中參考文獻)。Fig.1 The mechanism of PM2.5 affecting bone metabolism at low estrogen levels
1.3芳香烴受體(AhR)
芳香烴受體(AhR)廣泛分布于人體組織器官中(如骨),與ER以復雜的方式相互起干擾作用,且具有類雌激素或抗雌激素樣作用。TCDD、PAHs、BaP等是典型芳基烴受體(AhR)激動劑和CYP1A1誘導劑,并通過AHR與ERα之間相互干擾來共同競爭激活Arnt核蛋白,干擾雌激素信號傳導,促進破骨細胞基因表達[44]。多環芳烴(PAHs)介導 NF-kB誘導巨噬細胞和破骨細胞表達CYP1B1(一種雌激素代謝酶)增加,使骨中雌激素水平降低,導致骨成積率降低,影響骨重塑[45]。Iqbal等[46]指出,破骨細胞作用在Cyp1a1/1a2/1b1-/-三重KO小鼠骨吸收減少,并且在AhR-/-的小鼠,骨量顯著增加。H?d′alová等[47]利用雌激素敏感的人乳腺癌MCF-7細胞的AhR基因敲除變異體,觀察到芳香烴化合物及代謝物在MCF-7 AhRKO細胞中的雌激素樣作用減弱,而CYP1A1和CYP1B1酶的異位表達部分恢復了BAP代謝及其對細胞增殖的影響。另外,在對敲除AhR(RANKΔOc/ΔOc)的骨髓巨噬細胞培養中,也發現B淋巴細胞誘導CYP1B1、CYP1A2表達下調,呈現破骨功能受損和分化抑制[48]。
此外,芳香烴化合物通過激活AhR來抑制SMAD依賴的信號通路TGF-β1/Smad4和SMAD非依賴的TGF-β1/ERK/AKT信號通路,使其下游相應蛋白磷酸化降低,成骨細胞基因Runx2和OPN明顯降低,導致成骨分化和成熟均受抑制,骨生成減少[49]。同時,激活AhR還能通過RANK/c-Fos信號軸調控髓系前體向破骨細胞的分化,在AhR過表達的BMMS中,骨吸收面積和CTX-1的釋放均明顯升高,促進破骨作用[50]。激活AhR還可介導影響NF-κB、Wnt、MAPK等信號通路蛋白的轉錄促進破骨作用[51]。
2 其他介導途徑
垂體-骨軸具有內分泌骨重塑調節的功能。垂體激素(FSH)能通過破骨細胞及其前體物上的Gi 2α偶聯FSH受體(FSHR)來增強ERK、Akt、 IκBα(核因子κB-輕鏈增強子)的磷酸化,從而增強ERK/c-Fos和NF-κB信號轉導,介導RANKL的促骨吸收作用[52]。另外,FSH可直接刺激骨髓微環境產生TNFα、IL-1β、IL-6,而FSHβ基因缺陷小鼠骨髓巨噬細胞產生炎癥因子減少,即使雌激素缺乏,仍可免于骨量丟失[53-54]。PM2.5暴露誘導下丘腦炎癥因子釋放,從而抑制下丘腦-垂體-性腺軸,影響垂體LH和FSH釋放,影響相應功能[55]。應用FSH特異性抗體可增加骨量[56]。
3 結語
PM2.5可通過多途徑、多通路促進雌激素缺乏癥的骨質丟失。近年來,國內有關學者越來越注意到環境污染對骨侵蝕的作用。筆者有望通過在體水平或細胞水平建模探討PM2.5暴露對年齡相關性雌激素缺乏介導骨質丟失發生、發展的研究。
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
