
以中樞性尿崩癥為首發表現的肝豆狀核變性1例報告
2020-06-16 09:15:28趙茂,劉蕊,胡曉
臨床肝膽病雜志 2020年5期
關鍵詞:癥狀
趙 茂, 劉 蕊, 胡 曉
1 遵義醫科大學及其附屬醫院, 貴州 遵義 563003; 2 貴州省人民醫院, 貴陽 550001
肝豆狀核變性(hepatolenticular degeneration,HLD),又稱Wilson病,是一種常染色體隱性遺傳的銅代謝障礙性疾病[1],該病在世界范圍內的發病率為1/3萬~1/10萬[2],是至今少數早期診斷及正確治療可以得到較好療效的遺傳代謝性疾病之一。其主要發病機制為13號染色體上ATP7B基因純合或復合雜合突變,導致其編碼產物ATP7B的功能缺陷,引起血清銅藍蛋白合成減少及膽道排銅障礙,大量游離銅離子沉積于肝、腦、角膜等處造成相應器官損傷[1,3]。由于銅在不同部位的異常沉積,臨床表現多樣,最常見為肝損傷、錐體外系癥狀、角膜色素環[4]。以中樞性尿崩癥為首發癥狀的HLD罕見,容易延誤診斷,現將貴州省人民醫院收治的1例報道如下。
1 病例資料
患者男性,19歲,因“煩渴、多飲、多尿1個月余”于2017年10月8日就診貴州省人民醫院內分泌科。患者在1月余前感冒后出現煩渴、多飲、多尿。飲水量約10 L/d,尿量與飲水量相當,期間因口渴未能及時飲水出現畏寒、發熱,體溫最高達38.5 ℃,伴乏力。上述癥狀持續無好轉。既往史、家族史無特殊。入院查體:生命體征平穩,體型消瘦,面部可見蜘蛛痣1枚,雙下肢皮膚可見色素沉著。腹軟,無壓痛及反跳痛。肝肋下未觸及,脾左鎖骨中線和肋緣交點下6 cm可捫及。
入院后完善相關檢查,血常規:WBC 2.66×109/L,RBC 4.22×1012/L,Hb 118 g/L,PLT 54×109/L;肝功能:AST 102 U/L,GGT 123 U/L,ALP 227 U/L,總蛋白47.5 g/L,白蛋白22 g/L,球蛋白25.5 g/L,白球比值0.86。尿滲透壓49 mosm·kg-1·H2O-1,尿比重1.005,行禁水加壓素試驗提示中樞性完全性尿崩癥。腹部B超示:肝硬化并脾臟腫大、膽囊壁水腫;肝硬度示:脂肪衰減值171 dB/m, 肝硬度值12.2 kPa。上腹部增強CT示:肝硬化征象,脾大、腹水、門靜脈高壓、側支循環開放。請感染科會診后查:血清銅藍蛋白13.3 mg/dl,血清銅601.7 μg/L,24 h尿銅417.9 μg。頭顱MRI平掃示:腦白質散在缺血灶(Fazekas 1級),磁敏感加權成像未見異常。垂體MRI平掃及增強示:垂體后葉短T1高信號消失(圖1)。請眼科會診后行雙眼眼前節照相,雙眼角膜緣可見K-F環(圖2)。
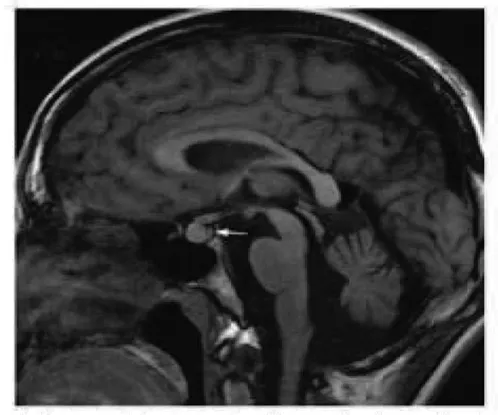
注:垂體后葉短T1高信號消失(箭頭所示)。
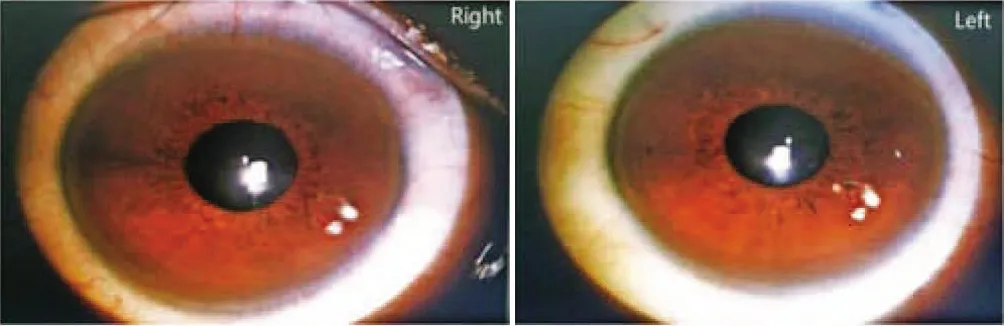
注:雙眼角膜緣可見K-F環。
根據患者癥狀體征及實驗室等檢查結果,考慮HLD可能性大,但患者除肝損傷外,尿崩癥癥狀突出,臨床表現不典型,為進一步確診,在患者及其家屬的知情同意下,對患者及其家屬進行基因檢測。采用Sanger測序法,對ATP7B基因21個外顯子及側翼序列進行突變分析,結果發現,該患者ATP7B基因存在兩個雜合致病突變:2299dupC(Met769Hisfs*26),2827G>A(Gly 943Ser)。其母親基因檢測為2299dupC單位點突變,單純雜合子。其父親基因檢測為2827G>A 單位點突變,單純雜合子。父母均無癥狀,尿銅正常。
最終診斷為HLD合并中樞性尿崩癥,予低銅飲食、青霉胺聯合硫酸鋅合劑驅銅、補鋅、醋酸去氨加壓素口服等對癥治療。經治療后,復查肝功能:AST 46 U/L,ALP 210 U/L,GGT 85 U/L;復查24 h尿銅:189 μg,同治療前相比呈下降趨勢,驅銅治療有效,患者臨床癥狀好轉,入院14 d后帶藥出院。隨訪1年半,患者多尿癥狀明顯減輕,但仍反復出現腹水等肝硬化癥狀,繼續治療中。
2 討論
HLD是一種銅代謝障礙所致疾病,為常染色體單基因隱性遺傳病。HLD患者多數以肝病和神經精神癥狀起病,少數以腎損害、骨關節損害及血液系統癥狀等起病[5]。由于該病常涉及多個器官系統,臨床表現復雜多樣,且病程進展不同,早期容易誤診及漏診[6-7]。本例患者以尿崩癥起病,雖有肝臟受損表現,但尿崩癥癥狀突出,禁水加壓素實驗及垂體MRI檢查,提示有中樞性尿崩癥,是否與HLD相關,值得進一步探討。
中樞性尿崩癥是一種較為少見的內分泌系統臨床綜合征,主要以多尿、煩渴、多飲、低比重尿及低滲透壓尿為特征。其發病機制主要為下丘腦神經垂體病變導致抗利尿激素分泌不足。本例患者以多尿、煩渴、多飲起病,垂體MRI及禁水加壓素實驗有中樞性尿崩癥特征,但既往無顱腦外傷及手術史,而且查頭顱CT未見異常鈣沉積高信號,頭顱MRI未見異常鐵沉積及占位病變,垂體MRI未見炎癥及腫瘤病變,可排除單純性尿崩癥、顱腦外傷及手術、垂體腫瘤及炎癥、異常鈣或鐵沉積導致垂體病變,推測中樞性尿崩癥為銅異常沉積所致。臨床進一步證實血清銅藍蛋白明顯下降、角膜K-F環,且患者存在無法解釋的肝硬化,說明體內存在銅代謝異常并累及多系統。最終經基因檢測證實為HLD。因此,考慮患者出現中樞性尿崩癥等內分泌紊亂是由HLD導致的。
目前尚無臨床案例報道,明確提及HLD表現為尿崩癥者。但既往臨床報道中有HLD患者出現垂體功能減退癥[8]、下丘腦-垂體-性腺軸功能損害[9-10]、多囊卵巢綜合征[11]等內分泌系統受累表現;與本例患者類似,因此推測患者不考慮單純性尿崩癥,而是HLD導致繼發性中樞性尿崩癥。由HLD導致中樞性尿崩癥發生的可能機制為銅代謝異常致垂體內分泌紊亂。由于銅離子在下丘腦垂體的沉積,破壞下丘腦、垂體細胞及亞細胞的結構和功能,使下丘腦-垂體功能缺陷或受損,導致垂體分泌抗利尿激素功能下降,產生中樞性尿崩癥。
HLD患者的影像學特點為病變范圍分布廣泛,而且由于疾病的不同發展時期和MRI系統的特性使病變表現具有多樣性[12]。既往研究[4]已表明,HLD患者頭顱MRI可發現豆狀核(尤其殼核)、尾狀核、中腦、腦橋、丘腦、小腦及額葉皮質Tl加權像低信號和T2加權像高信號;近期研究者[13-14]發現,應用磁共振彌散張量成像、靜息態功能磁共振、磁敏感成像等影像學檢查可檢測HLD患者腦部丘腦等核團存在結構性損傷及功能活動水平的下降。本例患者因多飲、多尿就診,頭顱MRI提示腦白質散在缺血灶(Fazekas 1級),磁敏感加權成像檢查未見明顯異常,非典型HLD影像學改變。為進一步確診,患者完善血清銅藍蛋白、角膜K-F環及ATP7B基因檢測,結果均支持HLD診斷。因此,對臨床疑似HLD患者,尤其頭顱MRI等影像學表現不典型患者,應盡早完善銅代謝生物化學、K-F環等特異性檢查,并結合基因檢測進行綜合判斷。
基因診斷是目前HLD早期確診的最有效方法[1],尤其對臨床癥狀、生化及影像學特點不典型患者有重要意義。國內外指南[4,15]也一致推薦,對任何臨床及生化檢查難以確定的疑似HLD均應進行基因檢測。ATP7B基因是目前已知唯一的HLD致病基因,但其突變位點及類型復雜,而且該基因突變具有種族差異[1],歐美HLD患者以p.His1069Gln突變最多見[16]。在我國最常見的突變類型為p.Arg778Leu,其次為p.Pro992Leu,相關研究[17]也表明我國HLD患者基因突變以少數幾個熱點突變和廣泛罕見突變為特征。HLD患者的基因水平確診需滿足ATP7B基因純合致病突變或復合雜合致病突變,單一雜合突變不能確診[18],一項對ATP7B基因突變位點的研究[19]也發現,復合突變者常比單一突變者更早發病。結合本例HLD患者,ATP7B 基因測序為p.Met769Hisfs*26和p.Gly 943Ser兩個位點的突變,為復合基因突變型,其父母均發生一種雜合突變,表型正常,故可確診患者因遺傳父母雙方的雜合突變而發生HLD。
綜上所述,對于中樞性尿崩癥患者,合并不明原因的肝臟病變需警惕HLD,臨床醫生應仔細詢問病史,同時要平行檢查血清銅藍蛋白、血銅、24 h尿銅、角膜K-F環、ATP7B基因等多項指標,請感染科、眼科等相關科室會診,以降低誤診率和漏診率,使患者得到早診斷、早期干預治療,最終改善疾病預后。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
