近紅外長(zhǎng)余輝納米探針ZnGa2O4∶Cr3+,Sn4+的制備及Fe3+含量的時(shí)間分辨檢測(cè)
2020-06-16 09:21:58靜邵康王鍇張聰滕淵潔潘再法佘遠(yuǎn)斌
發(fā)光學(xué)報(bào) 2020年6期
關(guān)鍵詞:檢測(cè)
林 靜邵 康王 鍇張 聰滕淵潔潘再法佘遠(yuǎn)斌
(浙江工業(yè)大學(xué)化學(xué)工程學(xué)院,浙江杭州 310000)
1 引 言
鐵是人體必需的微量元素之一[1],作為人體中重要的生命要素[2],在人體的新陳代謝過(guò)程中具有重要作用[3]。缺鐵會(huì)導(dǎo)致人體的免疫力降低,出現(xiàn)缺鐵性貧血。通常情況下,攝入鐵的主要方式有食補(bǔ)和補(bǔ)鐵保健品,其中服用補(bǔ)鐵口服液是最為常見(jiàn)的方法。補(bǔ)鐵口服液具有補(bǔ)氣補(bǔ)血的效果,可有效增強(qiáng)人體中的含氧量,改善氣血循環(huán)。但是過(guò)度攝入鐵則會(huì)導(dǎo)致組織損傷、器官衰竭甚至死亡[4],因此測(cè)定補(bǔ)鐵口服液中鐵元素的含量具有非常重要的意義。
常見(jiàn)鐵的測(cè)定方法有分光光度法[5]、原子吸收光譜法[6]、比色法[7]、熒光光譜法[8]等。相比于其他檢測(cè)方法,熒光光譜法具有操作簡(jiǎn)便、重復(fù)性好、靈敏度高等優(yōu)點(diǎn)[9-11],可實(shí)現(xiàn)Fe3+含量的簡(jiǎn)便快速檢測(cè)。至今為止,已有研究人員采用多種熒光材料構(gòu)建熒光探針用于檢測(cè)Fe3+的含量,但是大多檢測(cè)Fe3+含量的探針尚存在一些缺點(diǎn),如背景干擾大、選擇性不強(qiáng)或隨pH值變化影響等[12-14]。2017年,Jiang等合成一種星形三苯并噻唑基苯(TBB)作為熒光探針檢測(cè)水中的Fe3+,TBB的熒光性能受到pH的影響較大,在強(qiáng)堿性環(huán)境下具有良好的耐受性,但在強(qiáng)酸的條件下會(huì)發(fā)生質(zhì)子化,導(dǎo)致電荷轉(zhuǎn)移引起熒光變化,對(duì)Fe3+的測(cè)定會(huì)產(chǎn)生干擾[15]。2019年,Jayaweera等[16]采用榴蓮殼為原料制備熒光碳點(diǎn),用于檢測(cè)水體中的Fe3+,采用365 nm作為最佳激發(fā)波長(zhǎng),可產(chǎn)生藍(lán)色熒光,但無(wú)法實(shí)現(xiàn)免實(shí)時(shí)激發(fā)檢測(cè),未能有效避免激發(fā)光產(chǎn)生的背景干擾。Ye等[17]采用摻雜鑭系配位聚合物制備碳點(diǎn)構(gòu)建比率型熒光傳感用于自來(lái)水中Fe3+的定量檢測(cè)。目前檢測(cè)Fe3+含量的熒光探針大多無(wú)法避免激發(fā)光和復(fù)雜樣品自體熒光的干擾,常應(yīng)用于水樣或其他簡(jiǎn)單樣品的實(shí)際測(cè)定,在復(fù)雜樣品的Fe3+檢測(cè)方面仍需進(jìn)一步探究。因此,開(kāi)發(fā)新型熒光探針用于復(fù)雜樣品中Fe3+的快速準(zhǔn)確檢測(cè)具有重要意義[18]。
長(zhǎng)余輝材料又稱為夜光材料,在經(jīng)過(guò)外界光源激發(fā)之后,可將能量?jī)?chǔ)存在陷阱中,在停止激發(fā)后仍可持續(xù)發(fā)光,在安全顯示、光電儲(chǔ)存、儀表顯示以及生物成像等領(lǐng)域具有廣泛的應(yīng)用[19]。由于長(zhǎng)余輝材料在停止激發(fā)后,仍具有較強(qiáng)熒光,因此可做到免實(shí)時(shí)激發(fā)檢測(cè),有利于消除激發(fā)光源及復(fù)雜樣品自體熒光的干擾。另外,由于長(zhǎng)余輝材料具有獨(dú)特的余輝特性,不需要采用具有熒光時(shí)間分辨功能的昂貴儀器,就可實(shí)現(xiàn)磷光的時(shí)間分辨測(cè)定,可有效消除復(fù)雜樣品的自體熒光的干擾,實(shí)現(xiàn)無(wú)背景干擾下的高信噪比檢測(cè)[20-21]。2010年,嚴(yán)秀平等[22]首次利用 Ca1.86Mg0.14Zn-Si2O7∶Eu2+,Dy3+與金納米粒子結(jié)合,根據(jù)熒光共振能量轉(zhuǎn)移(FRET)機(jī)理構(gòu)建綠光長(zhǎng)余輝熒光探針,并成功應(yīng)用于血清樣品中甲胎蛋白(AFP)的檢測(cè),有效地避免了血清樣品的自體熒光和激發(fā)光的干擾,檢出限低至0.41 μg/L。2018年,Hu等[23]采用綠光長(zhǎng)余輝材料Sr2Al14O25∶Eu2+,Dy3+構(gòu)建熒光傳感用于抗生素和2,4,6-三硝基酚(TNP)的檢測(cè),該生物傳感器有效地避免了背景干擾并成功地應(yīng)用于牛奶、水樣中污染物的檢測(cè)。陳學(xué)元等[24]制備了近紅外長(zhǎng)余輝材料ZnGa2O4∶Cr3+,將其進(jìn)行生物素化后用于構(gòu)建熒光傳感體系,有效地避免了蛋白質(zhì)自體熒光的影響,可在白光激發(fā)下實(shí)現(xiàn)重復(fù)激發(fā),實(shí)現(xiàn)了親和素蛋白的異構(gòu)測(cè)定。
在長(zhǎng)余輝材料中,采用Cr3+摻雜的鎵酸鹽長(zhǎng)余輝材料具有良好的化學(xué)穩(wěn)定性,可在強(qiáng)酸強(qiáng)堿等環(huán)境下保持良好的發(fā)光性能,并在近紅外區(qū)域具有較強(qiáng)的熒光發(fā)射。由于Cr3+摻雜的鎵酸鹽長(zhǎng)余輝材料具有良好的余輝性能、化學(xué)穩(wěn)定性和組織透過(guò)性等優(yōu)點(diǎn)[25],在生物成像和熒光傳感領(lǐng)域具有廣泛的應(yīng)用前景,受到了廣大研究者的密切關(guān)注。因此本文采用水熱法合成Sn4+共摻的近紅外長(zhǎng)余輝材料ZnGa2O4∶Cr3+,Sn4+(ZGSC),并進(jìn)一步進(jìn)行包硅處理,以獲得良好的水中分散性。利用Fe3+對(duì)所合成的長(zhǎng)余輝納米探針的猝滅效應(yīng),建立了操作簡(jiǎn)便、選擇性強(qiáng)、干擾小的熒光傳感方法,實(shí)現(xiàn)補(bǔ)鐵口服液中Fe3+的含量高選擇性檢測(cè)。并且采用時(shí)間分辨光譜可有效地消除背景干擾,獲得高信噪比。市面上大多補(bǔ)鐵口服液的有效價(jià)態(tài)是二價(jià)鐵[26],但目前報(bào)道的通常是測(cè)定補(bǔ)鐵口服液中的總鐵含量。本文可以分別測(cè)定補(bǔ)鐵口服液中的總鐵含量和Fe3+的含量,并根據(jù)總鐵含量和Fe3+的含量計(jì)算出二價(jià)鐵的含量,具有分別檢測(cè)Fe3+與Fe2+的優(yōu)點(diǎn)。
2 實(shí) 驗(yàn)
2.1 長(zhǎng)余輝材料的合成
按照化學(xué)計(jì)量比稱取1 mmol Zn(CHCOO)2·2H2O、2 mmol Ga(NO3)3·xH2O、0.002 mmol Cr(CHCOO)3·2H2O及0.004 mmol SnCl4·5H2O溶于20 mL去離子水中,攪拌1 h。加入NaOH(2 mol/L)調(diào)節(jié)pH=11,繼續(xù)攪拌2 h后,將反應(yīng)物轉(zhuǎn)移至25 mL的水熱反應(yīng)釜中,反應(yīng)釜于200℃下反應(yīng)18 h,待冷卻后離心,并用去離子水和無(wú)水乙醇洗滌3次,沉淀物置于烘箱中在60℃下干燥12 h,得到水熱后的長(zhǎng)余輝納米材料。再將產(chǎn)物轉(zhuǎn)移至剛玉坩堝中,設(shè)置升溫速率為10℃/min,于1 000℃煅燒4 h,冷卻至室溫后,用研缽研磨,得到產(chǎn)物ZGSC。
稱取30 mg煅燒后的ZGSC,加入24 mL無(wú)水乙醇、0.1 mL硅酸四乙酯及1.44 mL氨水,攪拌反應(yīng)4 h。再轉(zhuǎn)移至離心管中離心,采用去離子水與無(wú)水乙醇各洗滌3次,沉淀物置于烘箱中于60℃下干燥12 h,得到在表面包覆上硅殼的長(zhǎng)余輝納米材料ZGSC@SiO2。
2.2 測(cè)定條件優(yōu)化及選擇性實(shí)驗(yàn)
采用HCl溶液(0.1 mol/L)和 NaOH溶液(0.1 mol/L)配制 pH 為 5,6,7,8,9,10,11,12,13的溶液,備用。分別取200 μL不同pH的溶液與100 μL ZGSC@SiO2混合均勻,測(cè)定混合溶液的熒光光譜,考察pH值對(duì)長(zhǎng)余輝探針熒光強(qiáng)度的影響。
配制濃度為 0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9,1,2,3,4,5 mg/mL 的 ZGSC@ SiO2水分散液,測(cè)定不同濃度ZGSC@SiO2水分散液的熒光光譜,考察ZGSC@SiO2水分散液濃度對(duì)長(zhǎng)余輝探針熒光強(qiáng)度的影響。
配制濃度為10-2mol/L的 K+、Na+、Mg2+、Al3+、Zn2+、Fe2+、Fe3+、Hg2+、Cu2+、Co2+、Cd2+、Cr3+、NH4+、Mn2+和 Ag+等金屬離子溶液備用。分別取200 μL的金屬離子溶液與100 μL ZGSC@SiO2混合均勻,測(cè)定混合溶液的熒光光譜,考察不同金屬離子對(duì)長(zhǎng)余輝探針熒光強(qiáng)度的影響。
2.3 Fe3+的定量測(cè)定及標(biāo)準(zhǔn)曲線制作
配制不同濃度的 Fe3+溶液,分別取100 μL ZGSC@SiO2與200 μL Fe3+溶液混合,攪拌1 min后,選擇254 nm為激發(fā)波長(zhǎng),測(cè)定混合物的發(fā)射光譜。以最大發(fā)射波長(zhǎng)695 nm處的熒光強(qiáng)度對(duì)Fe3+濃度作圖,得到Fe3+的標(biāo)準(zhǔn)曲線,用于實(shí)際樣品中的Fe3+濃度定量測(cè)定。
2.4 實(shí)際樣品檢測(cè)
選擇3種不同的市售補(bǔ)鐵口服液(10 mL/支)作為實(shí)際樣品,取適量體積的補(bǔ)鐵口服液樣品稀釋至50 mL使得待測(cè)樣品的濃度在Fe3+的線性范圍內(nèi),定容備用。分別取100 μL ZGSC@SiO2、100 μL 雙氧水與100 μL 待測(cè)樣品于比色皿中,超聲1 min混合均勻。其中補(bǔ)鐵口服液中的Fe2+可被雙氧水氧化為Fe3+,故可測(cè)定樣品中的總鐵含量。測(cè)定混合物的發(fā)射光譜,計(jì)算總鐵含量。 取 100 μL ZGSC@ SiO2、100 μL 去離子水與100 μL待測(cè)樣品混合均勻,超聲1 min,測(cè)量混合物的發(fā)射光譜,測(cè)定樣品中Fe3+的含量。計(jì)算總鐵含量和三價(jià)鐵含量的差值,可得樣品中Fe2+的含量。
2.5 儀器與測(cè)試
采用Fluoromax-4P熒光光譜儀(法國(guó)HORIBA Jobin Yvon公司)測(cè)定長(zhǎng)余輝材料的發(fā)射光譜,ZnGa2O4∶Cr3+,Sn4+的激發(fā)波長(zhǎng)為 254 nm,發(fā)射光譜的監(jiān)測(cè)波長(zhǎng)為695 nm。X'pert PRO X射線衍射儀(荷蘭PANlytical公司)用于測(cè)定樣品的X射線衍射譜。采用高分辨透射電子顯微鏡(荷蘭Tecnai G2 F30)分析樣品的形貌,加速電壓300 kV。
3 結(jié)果與討論
3.1 長(zhǎng)余輝材料的結(jié)構(gòu)表征
ZnGa2O4晶體具有立方尖晶石結(jié)構(gòu)(空間組Fd3m),Zn2+占據(jù)四面體格位(Td),占據(jù)八面體格位(D3d)。由于摻雜的Cr3+離子與Ga3+具有相同的原子價(jià)和相似的離子半徑(0.061 5 nm vs.0.062 nm),因此它們優(yōu)先占據(jù)晶體Ga3+位置[27]。同時(shí)摻雜 Cr3+與 Sn4+可有效地提升ZnGa2O4的余輝性能。采用X射線衍射儀(XRD)以及高分辨透射電子顯微鏡(TEM)對(duì)長(zhǎng)余輝材料的晶體結(jié)構(gòu)和尺寸形貌進(jìn)行表征分析。分別對(duì)1 000℃高溫煅燒后的ZGSC以及包硅之后的ZGSC@SiO2兩種樣品進(jìn)行了XRD表征,如圖1所示。兩種不同階段長(zhǎng)余輝材料的XRD圖譜均與ZnGa2O4的標(biāo)準(zhǔn)卡片相符。從圖1中可看出,經(jīng)過(guò)煅燒之后ZGSC的峰尖且窄,說(shuō)明經(jīng)過(guò)煅燒后樣品結(jié)晶性完好,包硅處理均未改變長(zhǎng)余輝材料的晶體結(jié)構(gòu)。圖2分別為水熱處理、高溫煅燒及包硅處理之后的ZGSC的TEM圖。

圖1 ZGSC和 ZGSC@SiO2的XRD圖,ZnGa2O4的標(biāo)準(zhǔn)卡片為PDF#86-0413。Fig.1 XRD of ZGSC and ZGSC@ SiO2.The standard card of ZnGa2O4is PDF#86-0413.

圖2 (a)~(b)ZGSC的透射電鏡圖;(c)1 000℃煅燒后ZGSC的透射電鏡圖;(d)包硅處理后ZGSC@SiO2的透射電鏡圖。Fig.2 (a)-(b)TEM and HRTEM images of ZGSC.(c)TEM of ZGSC after 1 000℃calcination.(d)TEM of ZGSC@SiO2.
圖2 (a)、(b)均為水熱處理之后長(zhǎng)余輝材料的TEM圖。從圖2(a)中可觀察到水熱反應(yīng)獲得的ZGSC粒徑較小,其平均粒徑均小于50 nm,且晶體粒徑較為均一。從圖2(b)中可觀察到水熱制得的ZGSC具有清晰的晶格條紋,其間距為0.158 nm,由此可說(shuō)明水熱反應(yīng)制得的ZGSC結(jié)晶性良好。圖2(c)為1 000℃煅燒后ZGSC的TEM圖,從圖中可觀察到,其晶體大小較為均勻,經(jīng)過(guò)煅燒后晶體粒徑有所增大,但其平均粒徑均小于200 nm。圖2(d)為包硅處理后ZGSC@SiO2的TEM圖,從圖中可觀察到在ZGSC表面包覆了一層明顯的硅層,經(jīng)過(guò)包硅之后,ZGSC表面修飾上大量的硅羥基Si—OH,因此ZGSC的親水性得到有效地提高,從而提升了ZGSC在水相中的分散性。并且經(jīng)過(guò)包硅處理,ZGSC@SiO2可在水相中穩(wěn)定分散1 h以上,由此說(shuō)明包硅效果良好。
3.2 熒光性能表征
長(zhǎng)余輝材料的激發(fā)發(fā)射光譜如圖3所示,從激發(fā)光譜圖中可觀察到,通過(guò)監(jiān)測(cè)695 nm處的發(fā)射,在紫外-可見(jiàn)區(qū)存在以254,369,497 nm 為中心的3個(gè)寬激發(fā)帶。其中在254 nm處有一強(qiáng)的激發(fā)帶可歸因于帶間躍遷(CB→VB)及Cr3+的4A2(4F)→4T1(4P)躍遷,在369 nm和497 nm處的兩個(gè)峰分別是由于Cr3+的4A2(4F)→4T1(4F)和4A2(4F)→4T2(4F)的躍遷產(chǎn)生。從發(fā)射光譜圖中可看出,在254 nm激發(fā)下,可觀察到600~800 nm的近紅外區(qū)有一個(gè)寬的發(fā)射帶,其峰值位于695 nm,可歸因于2E→4A2躍遷。

圖3 ZGSC@SiO2的激發(fā)和發(fā)射光譜Fig.3 Excitation and emission spectra of ZGSC@ SiO2
除了激發(fā)光譜和發(fā)射光譜之外,還測(cè)定了ZGSC@SiO2的衰減曲線和時(shí)間分辨光譜(TR)。如圖4所示,采用254 nm作為激發(fā)光源,對(duì)長(zhǎng)余輝材料進(jìn)行充能,5 min之后關(guān)閉激發(fā)光源,300~600 s為衰減過(guò)程。首先是快速衰減過(guò)程,激發(fā)停止之后產(chǎn)生快速衰減的原因是被較淺的陷阱所捕獲的電子快速釋放;隨后是慢速衰減過(guò)程,慢速衰減是由于較深陷阱中的電子被緩慢釋放所導(dǎo)致。從圖4中可看出,經(jīng)過(guò)包硅處理后,ZGSC@SiO2在激發(fā)后所存儲(chǔ)的能量增大,且衰減速度沒(méi)有變快。由此可看出,包硅處理可有效地提高ZGSC的余輝性能。

圖4 ZGSC和ZGSC@SiO2的衰減曲線Fig.4 Decay curves of ZGSC and ZGSC@ SiO2
為了消除背景熒光的干擾,本文還測(cè)定了ZGSC@SiO2在 I=I0/e時(shí)(取 300 μs)的時(shí)間分辨光譜(TR),結(jié)果如圖5所示。從圖5中可以看出,在600~650 nm處,穩(wěn)態(tài)光譜基線明顯比時(shí)間分辨光譜的基線高,這是由于激發(fā)光源存在下的背景干擾。以695 nm處的熒光信號(hào)強(qiáng)度除以600 nm處的背景噪音強(qiáng)度,通過(guò)計(jì)算可得到穩(wěn)態(tài)下信噪比為5.9,時(shí)間分辨光譜中信噪比為42.7。可見(jiàn)利用時(shí)間分辨光譜,信噪比大幅提高,可有效地消除背景干擾,在時(shí)間分辨生物傳感等領(lǐng)域具有廣泛的應(yīng)用前景。
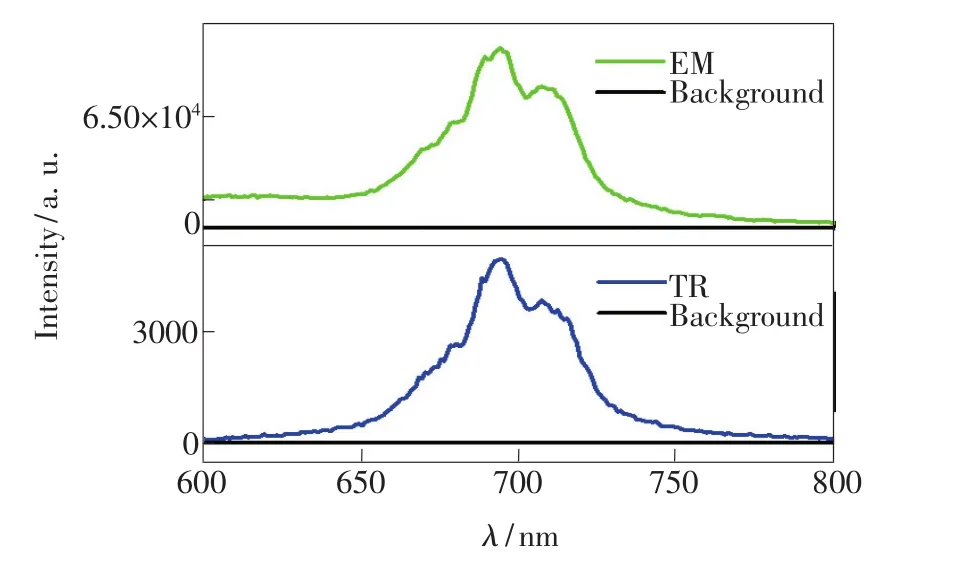
圖5 ZGSC@SiO2在穩(wěn)態(tài)和時(shí)間分辨的發(fā)射光譜Fig.5 Steady state and time resolved emission spectra of ZGSC@SiO2
3.3 測(cè)定條件優(yōu)化及選擇性實(shí)驗(yàn)

圖6 (a)ZGSC@SiO2在不同pH值環(huán)境中的熒光強(qiáng)度;(b)不同濃度ZGSC@SiO2水分散液的熒光強(qiáng)度。Fig.6 (a)Fluorescence intensity diagram of ZGSC@ SiO2in different pH.(b)Fluorescence intensity of ZGSC@SiO2water dispersion with different concentrations.
使用長(zhǎng)余輝材料ZGSC@SiO2作為熒光探針,其熒光強(qiáng)度會(huì)受到環(huán)境中pH值以及ZGSC@SiO2在水分散液中濃度的影響,故需對(duì)以上兩種因素進(jìn)行考察優(yōu)化。為探究環(huán)境中不同pH值是否會(huì)對(duì)ZGSC@SiO2熒光強(qiáng)度產(chǎn)生影響,本文分別測(cè)定了pH=5~13的環(huán)境下ZGSC@SiO2的熒光強(qiáng)度,結(jié)果如圖6(a)所示。在不同的pH環(huán)境中,ZGSC@SiO2的熒光強(qiáng)度基本保持不變,不同pH值并不會(huì)對(duì)ZGSC@SiO2的熒光性能產(chǎn)生較大影響,說(shuō)明ZGSC@SiO2具有良好的pH穩(wěn)定性,因此ZGSC@SiO2可用于廣泛pH范圍內(nèi)的檢測(cè)。
為探究分散在水溶液中長(zhǎng)余輝材料的濃度對(duì)熒光強(qiáng)度的影響,我們測(cè)定了不同濃度ZGSC@SiO2的熒光發(fā)射光譜,取695 nm處的熒光強(qiáng)度進(jìn)行對(duì)比,結(jié)果如圖6(b)所示。隨著ZGSC@SiO2濃度的不斷增大,其熒光強(qiáng)度呈現(xiàn)先增大后穩(wěn)定的趨勢(shì)。當(dāng)濃度為0.6 mg/mL時(shí),ZGSC@SiO2的熒光強(qiáng)度達(dá)到最大值;當(dāng)濃度大于0.6 mg/mL時(shí),ZGSC@SiO2的熒光強(qiáng)度逐漸趨于穩(wěn)定。因此,選擇0.6 mg/mL作為ZGSC@SiO2測(cè)定的最佳濃度。
除此之外,為探究近紅外長(zhǎng)余輝材料ZGSC@SiO2檢測(cè)Fe3+的可行性,我們考察了本傳感系統(tǒng)的選擇性。分別配制了濃度為10-2mol/L的不同金屬離子溶液,加入一定量的ZGSC@SiO2水分散液中混合,測(cè)定其熒光強(qiáng)度,從而探究常見(jiàn)陽(yáng)離子 K+、Na+、Mg2+、Al3+、Zn2+、Fe2+、Cu2+、Cr3+、Co2+、NH4+、Mn2+、Ag+、Cd2+對(duì) ZGSC@SiO2熒光性能的影響,實(shí)驗(yàn)結(jié)果如圖7所示。從圖7中可看出,加入其他常見(jiàn)陽(yáng)離子后,ZGSC@SiO2熒光強(qiáng)度沒(méi)有變化或變化不大,僅有Fe3+可使ZGSC@SiO2熒光強(qiáng)度產(chǎn)生大幅猝滅的現(xiàn)象。加入濃度為10-2mol/L的Fe3+之后,長(zhǎng)余輝材料ZGSC@SiO2的熒光被大幅猝滅,其猝滅效率達(dá)到了96.72%。

圖7 加入不同陽(yáng)離子后ZGSC@SiO2的熒光強(qiáng)度圖Fig.7 Fluorescence intensity of ZGSC@ SiO2in the presence of various ions
由此表明,長(zhǎng)余輝材料ZGSC@SiO2對(duì)Fe3+具有良好的選擇性,可用于實(shí)際樣品中Fe3+含量的選擇性檢測(cè)。Fe3+可使ZGSC@SiO2產(chǎn)生熒光猝滅的原因可能是由于長(zhǎng)余輝材料與Fe3+之間產(chǎn)生了光電子轉(zhuǎn)移。即由于ZGSC@SiO2的表面含有大量的羥基,加入Fe3+后,ZGSC@SiO2表面的羥基與Fe3+發(fā)生配位反應(yīng),可能導(dǎo)致激子的非輻射復(fù)合概率增加,從而產(chǎn)生猝滅效應(yīng)[28-31]。
3.4 Fe3+的定量測(cè)定及標(biāo)準(zhǔn)曲線制作
基于Fe3+可使ZGSC@SiO2產(chǎn)生熒光猝滅的現(xiàn)象,本研究建立了一種快速檢測(cè)Fe3+的傳感方法。為了探究Fe3+溶液的濃度與ZGSC@SiO2熒光強(qiáng)度之間的關(guān)系,向ZGSC@SiO2中加入濃度為0~10-2mol/L的Fe3+溶液后,測(cè)定了ZGSC@SiO2在254 nm激發(fā)時(shí)的發(fā)射光譜,如圖8(a)所示。從圖中可以看出,隨著 Fe3+的濃度在0~10-2mol/L之間不斷地增大,ZGSC@SiO2的熒光強(qiáng)度逐漸減弱,猝滅程度逐漸增大。選取695 nm處的熒光強(qiáng)度與Fe3+的濃度繪制標(biāo)準(zhǔn)曲線,結(jié)果如圖8(b)所示。在50~800 μmol/L之間,熒光強(qiáng)度與Fe3+之間具有良好的線性關(guān)系,線性方程為y= -6.4×104×lgC-1.9×105(R2=0.998 5),其中y為ZGSC@SiO2的熒光強(qiáng)度,lgC為Fe3+濃度的對(duì)數(shù)。重復(fù)測(cè)定3次以上,200 μmol/L Fe3+的相對(duì)標(biāo)準(zhǔn)偏差(RSD)為2.18%,檢出限為25.12 μmol/L。因此,本文構(gòu)建的熒光傳感體系可用于Fe3+的檢測(cè)。

圖8 (a)與不同濃度Fe3+溶液混合后ZGSC@SiO2的發(fā)射光譜;(b)檢測(cè)Fe3+的標(biāo)準(zhǔn)曲線。Fig.8 (a)Fluorescence emission spectra of ZGSC@ SiO2in the presence of Fe3+with different concentration.(b)Relationship between the fluorescence intensity of ZGSC@SiO2and concentration of Fe3+.
3.5 實(shí)際樣品的測(cè)定
為驗(yàn)證采用長(zhǎng)余輝材料ZGSC@SiO2用于檢測(cè)Fe3+含量的可行性,本文選用了市面上3種常見(jiàn)市售補(bǔ)鐵口服液作為實(shí)際樣品,樣品為1種乳酸亞鐵口服液和2種鈣鐵鋅口服液。分別測(cè)定了與不同實(shí)際樣品混合之后,ZGSC@SiO2的穩(wěn)態(tài)發(fā)射光譜以及關(guān)閉激發(fā)光源后I=I0/e(取300 μs)時(shí)的時(shí)間分辨光譜,結(jié)果如圖9(a)、(b)所示。從圖9(a)、(b)中可看出,隨著樣品中Fe3+的濃度增加,ZGSC@SiO2的熒光猝滅程度均逐漸增大。分別對(duì)兩種光譜的信噪比進(jìn)行計(jì)算,穩(wěn)態(tài)下的信噪比為5.3,時(shí)間分辨光譜中的信噪比為33.4,說(shuō)明采用時(shí)間分辨技術(shù)檢測(cè)鐵含量的信噪比明顯提高。通過(guò)圖9(a)與(b)的對(duì)比可看出,時(shí)間分辨光譜有效地消除了背景干擾,實(shí)現(xiàn)了免實(shí)時(shí)激發(fā)檢測(cè)。

圖9 (a)加入不同樣品時(shí)的ZGSC@SiO2的穩(wěn)態(tài)發(fā)射光譜;(b)與不同樣品混合后的ZGSC@SiO2的時(shí)間分辨光譜。Fig.9 (a)Emission spectra of ZGSC@ SiO2in different samples.(b)Time-resolved spectra of ZGSC@SiO2 in different samples.
為了驗(yàn)證所構(gòu)建的傳感體系的實(shí)用性,本研究利用ZGSC@SiO2/Fe3+傳感體系檢測(cè)了3種實(shí)際樣品中的總鐵含量和Fe3+含量,并進(jìn)行了加標(biāo)實(shí)驗(yàn)。實(shí)驗(yàn)結(jié)果如表1、表2所示。表1為實(shí)際樣品中的總鐵含量和Fe3+含量的測(cè)定結(jié)果。從表1中可看出,本方法測(cè)得的實(shí)際樣品中總鐵含量與標(biāo)注的總鐵含量較為相符,且實(shí)驗(yàn)結(jié)果表明實(shí)際樣品中Fe3+的含量較高,均大于總鐵含量的60%。實(shí)際樣品中總鐵含量的RSD為2.416%~3.808%,F(xiàn)e3+含量的RSD為3.263%~4.296%。實(shí)驗(yàn)結(jié)果證明,采用本研究構(gòu)建的熒光探針檢測(cè)實(shí)際樣品中Fe3+含量具有良好的重現(xiàn)性。
對(duì)實(shí)際樣品中的總鐵含量以及Fe3+含量分別進(jìn)行了加標(biāo)實(shí)驗(yàn),實(shí)驗(yàn)結(jié)果如表2所示。從表2中可看出,3種實(shí)際樣品中總鐵含量的平均加標(biāo)回收率為99.00%~99.79%,F(xiàn)e3+含量的平均加標(biāo)回收率為99.90%~102.69%,測(cè)定結(jié)果符合測(cè)定要求,說(shuō)明采用ZGSC@SiO2/Fe3+傳感體系檢測(cè)Fe3+的含量是可行的。除此之外,還可根據(jù)所測(cè)得的總鐵含量以及Fe3+的含量計(jì)算出實(shí)際樣品中的有效組分Fe2+含量,且實(shí)際樣品中Fe2+的含量較少,因此可說(shuō)明在補(bǔ)鐵口服液的生產(chǎn)、運(yùn)輸及儲(chǔ)存過(guò)程中,F(xiàn)e2+被大量氧化成Fe3+。可見(jiàn)本方法也可用于補(bǔ)鐵口服液中有效價(jià)態(tài)Fe2+的質(zhì)量控制檢測(cè)。
通常大多數(shù)采用熒光分析法檢測(cè)Fe3+都需要實(shí)時(shí)激發(fā),而常見(jiàn)的補(bǔ)鐵口服液的成分較為復(fù)雜,測(cè)定時(shí)存在強(qiáng)本底干擾,無(wú)法避免激發(fā)光和自體熒光的干擾[8]。與常見(jiàn)的補(bǔ)鐵試劑中Fe3+含量的檢測(cè)方法相比,本方法的優(yōu)勢(shì)是可同時(shí)檢測(cè)Fe3+和Fe2+含量,操作簡(jiǎn)單,簡(jiǎn)化了前處理過(guò)程。并且由于長(zhǎng)余輝材料具有獨(dú)特的余輝性能,可采用時(shí)間分辨技術(shù)實(shí)現(xiàn)免實(shí)時(shí)激發(fā)檢測(cè),避免激發(fā)光造成的干擾,對(duì)于成分復(fù)雜的補(bǔ)鐵藥品采用時(shí)間分辨技術(shù)可有效避免其本底干擾,有利于補(bǔ)鐵藥品中鐵含量的檢測(cè)。
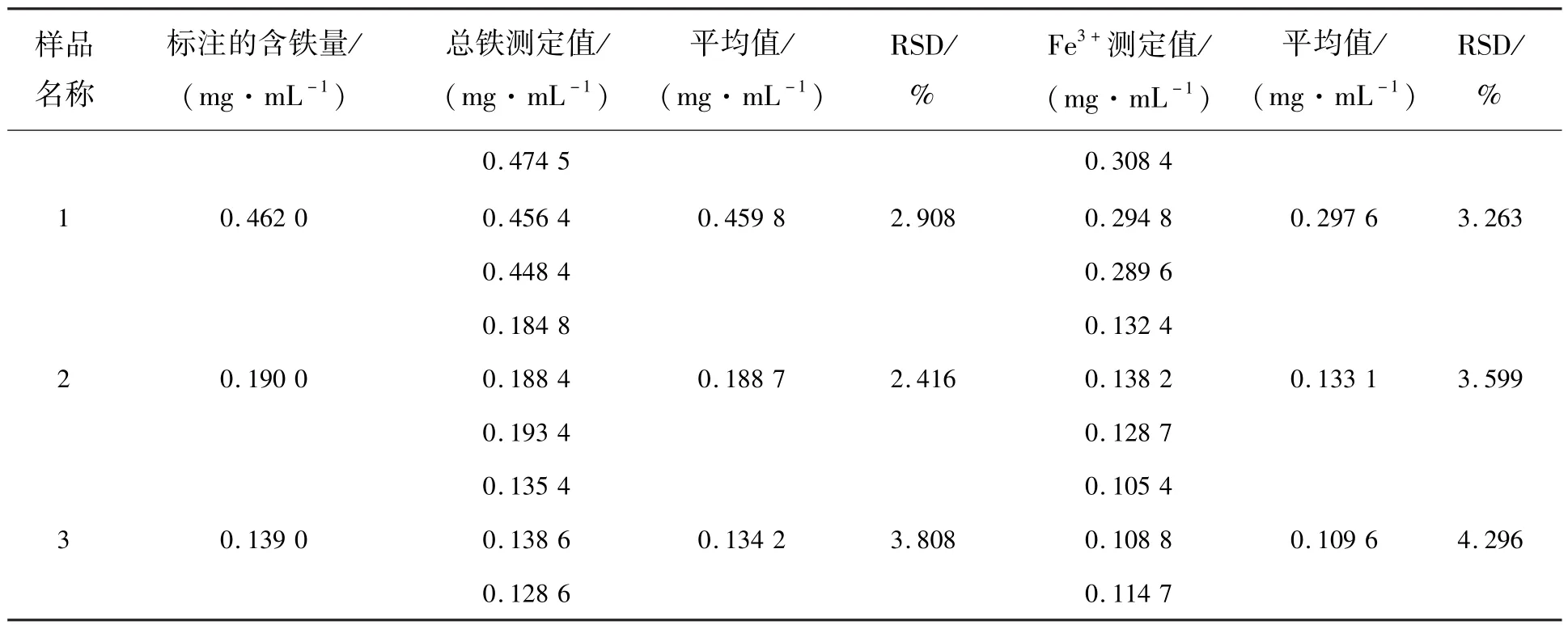
表1 樣品中總鐵含量和Fe3+含量的測(cè)定結(jié)果Tab.1 Determination results of total iron content and Fe3+in the samples

表2 樣品中總鐵含量和Fe3+含量加標(biāo)回收的測(cè)定結(jié)果Tab.2 Recovery test results of total iron content and Fe3+in the sample
4 結(jié) 論
本研究采用水熱法合成了納米級(jí)的近紅外長(zhǎng)余輝材料 ZnGa2O4∶Cr3+,Sn4+,并對(duì)其進(jìn)行了表面包硅處理得到ZGSC@SiO2,有效提高了長(zhǎng)余輝材料在水中的分散性。對(duì)長(zhǎng)余輝材料的晶體結(jié)構(gòu)和余輝性能進(jìn)行表征分析,考察了其余輝特性。基于Fe3+可使長(zhǎng)余輝材料ZGSC@SiO2發(fā)生熒光猝滅現(xiàn)象,本文構(gòu)建了新型熒光傳感體系用于Fe3+含量的檢測(cè),并實(shí)現(xiàn)了對(duì)3種補(bǔ)鐵口服液樣品中總鐵含量的檢測(cè)以及Fe3+含量的檢測(cè)。與其他檢測(cè)方法相比,本方法可通過(guò)測(cè)定實(shí)際樣品中的總鐵含量以及Fe3+含量,實(shí)現(xiàn)Fe3+與Fe2+的同時(shí)檢測(cè),可應(yīng)用于補(bǔ)鐵口服液中有效價(jià)態(tài)Fe2+的質(zhì)量控制。本方法具有信噪比高、無(wú)背景干擾、操作簡(jiǎn)便等優(yōu)點(diǎn)。并且采用時(shí)間分辨技術(shù)測(cè)定實(shí)際樣品中的鐵含量,有效地避免了激發(fā)光的背景干擾以及復(fù)雜樣品中的本底干擾,在保健品及復(fù)雜的生物樣品中鐵含量的檢測(cè)方面都具有廣泛的應(yīng)用前景。
猜你喜歡
中國(guó)設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
